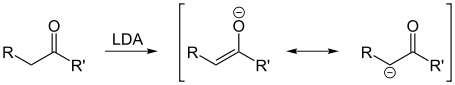
Enolate
Ein Enolat ist das Anion der Enolform einer Carbonylverbindung. Enolate entstehen durch die Deprotonierung eines CH-aciden Wasserstoffs in α-Position zur Carbonylfunktion. Es lassen sich zwei Grenzstrukturen formulieren, Enolate sind somit ambidente Anionen. Enolate sind gute Nucleophile, die mit weichen Elektrophilen (insbesondere Kohlenstoff-Elektrophilen) bevorzugt an der α-Position reagieren.
Erzeugung
Enolate entstehen durch Deprotonierung einer Carbonyl-Verbindung mit starken Basen. Zur unvollständigen Deprotonierung im Gleichgewicht reichen bereits Hydroxide oder Alkoholate aus (pKa von Ketonen ist ca. 20, von Estern ca. 25). Mit stärkeren Basen erreicht man eine vollständige Deprotonierung. Hierzu wird am häufigsten Lithiumdiisopropylamid (LDA), aber auch Lithiumhexamethyldisilazid (LHMDS), Kaliumhexamethyldisilazid (KHMDS) oder Lithiumtetramethylpiperidid (LTMP) verwendet. Allerdings dürfen die verwendeten Basen nicht selber nucleophil sein, da sie ansonsten die Carbonylverbindung am elektrophilen Carbonyl-Kohlenstoff angreifen und nicht deprotonieren. Daher sind beispielsweise Lithiumalkylverbindungen wie Butyllithium nicht geeignet.
Struktur
Regioselektivität
Bei asymmetrischen Ketonen sind oft zwei Regioisomere denkbar, die bei der Deprotonierung entstehen.
Unter kinetischer Kontrolle (starke Basen wie LDA, tiefe Temperaturen) wird bevorzugt das sterisch leichter zugängliche Proton entfernt, es entsteht die niedriger substituierte Enolat-Doppelbindung (kinetisches Enolat, 1).[1]
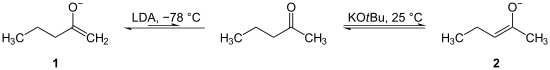
Unter thermodynamischer Kontrolle (schwache Base, höhere Temperatur) wird allerdings bevorzugt an der Stelle deprotoniert, wo mit Elektrophilen die höher substituierte Doppelbindung entstehen kann (thermodynamisches Enolat, 2).[1]
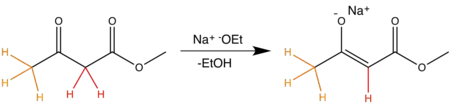
Die obere Abbildung zeigt ein weiteres Beispiel der thermodynamischen Kontrolle der Enolatbildung. Das stabilere Produkt ist das höher substituierte Enolat. Die Acidität der Wasserstoffsubstituenten ist hier durch verschiedene Farben gekennzeichnet, da es sich bei Natriumethanolat um eine relativ schwache Base handelt werden bevorzugt acidere Wasserstoffsubstituenten abstrahiert. Die orangefarbenen Wasserstoffsubstituenten weisen einen pka-Wert von ca. 20, die rot gefärbten Wasserstoffsubstituenten einen pka-Wert von ca. 12 auf.
Regioselektivität der Bildung von Enolaten aus Ketonen:[1]
| Thermodynamische Enolate | Kinetische Enolate |
|---|---|
| sind stärker substituiert | sind weniger stark substituiert |
| sind stabiler | sind weniger stabil |
| werden durch eine höhere Konzentration des Ketons, hohe Temperaturen und lange Reaktionszeiten begünstigt | werden durch starke, sterisch gehinderte Basen (z. B. LDA), niedrige Temperaturen und kurze Reaktionszeiten begünstigt. |
Stereoselektivität
Die Geometrie der Enolat-Doppelbindung (falls R > Methyl) wird durch die Größe des Substituenten R′ bestimmt. Es gibt hierfür zwei Möglichkeiten: (O)-(Z)-Enolate und (O)-(E)-Enolate. Der Zusatz (O) vor der E/Z-Angabe soll verdeutlichen, dass nur die Relativkonfiguration zwischen dem Enolat-Sauerstoff und dem Substituenten der Enolat-Doppelbindung betrachtet wird, was nicht mit der IUPAC-Nomenklatur für Doppelbindungen übereinstimmen muss. Große Substituenten R′ bevorzugen das (O)-(Z)-Enolat, kleine Substituenten das (O)-(E)-Enolat. Als große Substituenten kommen tertiäre Alkylsubstituenten sowie –NR2 in Frage. Als kleine Substituenten fasst man primäre Alkylsubstituenten sowie –OR und –SR auf.
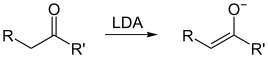 (O)-(Z)-Enolat, R′ = tertiär Alkyl, –NR2
(O)-(Z)-Enolat, R′ = tertiär Alkyl, –NR2
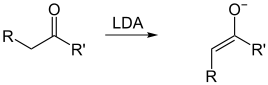 (O)-(E)-Enolat, R′ = primär Alkyl, –OR, –SR
(O)-(E)-Enolat, R′ = primär Alkyl, –OR, –SR
In der organischen Synthese werden chirale Auxilare zur stereoselektiven Reaktionsführung u. a. von Enolaten verwendet.
Das Ireland-Modell
Die Stereoselektivität der Enolatbildung kann hinreichend gut durch das Ireland-Modell erklärt werden.
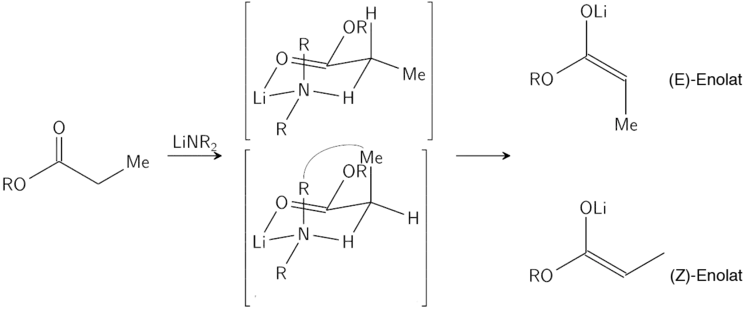
Im Ireland-Modell wird die Deprotonierung als ein sechsgliedriger Übergangszustand beschrieben. Der Größere der beiden Substituenten des Elektrophils (im oberen Fall ist die Methylgruppe größer, als der Wasserstoffsubstituent) bevorzugt dabei die äquatoriale Ausrichtung im Übergangszustand, um energetisch ungünstigen Wechselwirkungen mit den Substituenten der Base zu entgehen. Es wird daher hier das (E)-Enolat bevorzugt.
Das Modell versagt an vielen Punkten (z. B. wenn das Lösungsmittel von THF zu 23 % HMPA-THF verändert wird), es bietet aber eine gute Möglichkeit um eine Abschätzung der Stereoselektivität vorzunehmen.
Reaktionen
Als gute Nucleophile können Enolate mit einer Vielzahl von Elektrophilen umgesetzt werden. Mögliche Elektrophile sind unter anderem Alkylhalogenide (Alkylierung), Carbonsäurechloride (Acylierung), Carbonylverbindungen (Aldolreaktion), Michael-Akzeptoren, Epoxide, Vinyl- und Aryl-Halogenide. Alle diese Elektrophile greifen ausschließlich am C-Atom der Enolate an. Ein Angriff auf den Enolat-Sauerstoff ist selten und erfolgt nur durch harte Elektrophile (HSAB-Prinzip) wie beispielsweise Silylchloride oder Sulfonsäurederivate.
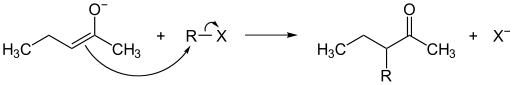
Literatur
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage, Spektrum Akademischer Verlag, München 2004, S. 516 ff., ISBN 3-8274-1579-9.
Einzelnachweise
- Jonathan Clayden, Nich Greeves, Stuart Warren: Organische Chemie. 2. Auflage. Springer Spektrum, Berlin / Heidelberg 2013, ISBN 978-3-642-34715-3, S. 659 ff.