Hämochromatose
Die Hämochromatose (von altgriechisch αἷμα haīma, deutsch ‚Blut‘ und altgriechisch χρῶμα chrōma, deutsch ‚Farbe‘; Synonyme: Primäre Siderose, Hämosiderose, Siderophilie, Eisenspeicherkrankheit, Bronzediabetes; englisch: iron overload, hemochromatosis) ist eine Erkrankung, bei der es durch eine erhöhte Aufnahme von Eisen im oberen Dünndarm und exzessive Eisenspeicherung (Einlagerung) vor allem in parenchymatösen Organen zu entsprechenden Organschäden kommt. Der Gesamtkörpereisengehalt steigt dadurch von ca. 2–6 g (Normwert) auf bis zu 80 g. Die Einlagerung führt im Laufe der Jahre zu Schädigungen insbesondere von Leber, Bauchspeicheldrüse, Herz, Gelenken, Milz, Hirnanhangsdrüse, Schilddrüse und Haut.
In den meisten Fällen handelt es sich bei der Hämochromatose um eine Erbkrankheit, die gewöhnlich autosomal-rezessiv vererbt wird. In diesem Fall müssen beide Eltern das veränderte Gen vererben, sie müssen aber nicht selbst die Krankheit haben (rezessiver Erbgang). Das für die Hämochromatose verantwortliche veränderte Gen liegt nicht auf einem Geschlechtschromosom (autosomaler Erbgang). In selteneren Fällen ist die Hämochromatose nicht erblich bedingt. Bei Männern hat die Hämochromatose eine wesentlich höhere Auftrittswahrscheinlichkeit als bei Frauen. Die Erkrankung kann, wenn sie frühzeitig entdeckt wird, erfolgreich behandelt werden. Bei fortgeschrittener Erkrankung treten unumkehrbare Schäden, insbesondere an der Leber, auf. Ebenso erhöht die Erkrankung das Risiko für die Entwicklung eines Leberzellkarzinoms.
| Klassifikation nach ICD-10 | |
|---|---|
| E83.1 | Störungen des Eisenstoffwechsels Hämochromatose |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Die Krankheit ist nicht meldepflichtig, daher liegen keine genauen Zahlen vor. Schätzungen gehen davon aus, dass in Deutschland über 200.000 Menschen mit einer Hämochromatose leben.[1]
Das HFE-Gen ist häufig von Mutationen betroffen, rund 10 % der nordeuropäischen Bevölkerung sind heterozygot (mischerbig) für einen solchen Gendefekt. Rund 0,3–0,5 % dieser Bevölkerungsgruppe sind hierfür homozygot (reinerbig), nur bei diesen kann die Krankheit auftreten. Die Penetranz ist aber unvollständig: Rund ein Drittel bis die Hälfte der homozygoten Mutationsträger zeigt keine klinischen Zeichen einer Eisenüberladung. Die verschiedenen Mutationen sind regional unterschiedlich verteilt. So wurden zahlreiche, in der westlichen Bevölkerung nicht vorkommende Mutationen bei Menschen asiatischer Abstammung nachgewiesen.[2] Hämochromatosen, die nicht mit einer HFE-Mutation einhergehen, sind sehr selten.[3] Sie treten gehäuft in Italien auf.[4]
Frauen verlieren natürlicherweise durch die Menstruation und bei Schwangerschaften im Körper gebundenes Eisen. Infolgedessen erkranken genetisch betroffene Männer fünf bis zehnmal häufiger. Die Latenzzeit bis zum Auftreten erster Krankheitszeichen variiert je nach Alkoholkonsum, Eisengehalt der Nahrung und Anzahl der erhaltenen Blutspenden. Die Mehrzahl der Erkrankten entwickelt die ersten Symptome zwischen dem vierten und sechsten Lebensjahrzehnt.[3]
Krankheitszeichen
Leberschäden, Diabetes, Hautpigmentierung
Die häufigsten Symptome der Hämochromatose sind neben einer Lebervergrößerung der Ausbruch eines Diabetes mellitus und eine dunkle Hautpigmentierung, weshalb die Hämochromatose gelegentlich auch als Bronzediabetes bezeichnet wird. Die Hautpigmentierung ist in der Achselhöhle meist am stärksten ausgeprägt. An den pigmentierten Stellen fehlen typischerweise auch die Körperhaare. Weitere bevorzugte Stellen der Hautverfärbungen sind die Streckseiten der Arme und Hände, der Hals- und Gesichtsbereich, die Unterschenkel sowie die Genitalregion. Drei Viertel der Patienten weisen bei Diagnosestellung bereits eine Leberzirrhose auf.[5][3]
Milzvergrößerung, Gelenkentzündung, Vorhofflimmern
Seltenere Krankheitszeichen sind Milzvergrößerung und die entzündliche Schwellung der Fingergrundgelenke. Die Gelenkentzündung kann im Verlauf auch auf größere Gelenke übergreifen. Sie läuft in vielen Fällen den anderen Symptomen zeitlich voraus. Rund 15 % der Erstdiagnosen werden erst durch Herzrhythmusstörungen auffällig. Möglich sind anfallsartiges Herzrasen, Vorhofflattern, Vorhofflimmern und Blockaden der Überleitung zwischen Vorhof und Kammer.
Psychische Krankheitszeichen und Hodenveränderungen
Als psychische Krankheitszeichen können Lethargie und Libidoverlust auftreten. Ebenso kann durch die Schädigung der Hirnanhangsdrüse ein Hypogonadismus auftreten. Zu den wichtigsten Allgemeinerkrankungen, die funktionelle und organische Hodenveränderungen (in der Regel Störungen der Spermiogenese) hervorrufen, gehören neben Kachexie (krankhafte, sehr starke Abmagerung) auch:[6]
- Leberzirrhose: Eine verminderte Östrogeninaktivierung als Ursache einer Hodenatrophie (umgangssprachlich „Schrumpfhoden“)
- Hämochromatose: Eisenablagerungen in der Hypophyse führen zu einem Panhypopituitarismus und somit zu einem sekundären Hypogonadismus (Testosteronmangel).[6] Als Panhypopituitarismus bezeichnet man eine Krankheit, bei der ein Mangel oder ein Fehlen sämtlicher in dem Hypophysenvorderlappen gebildeter Hormone besteht.
Einteilung der Hämochromatose
Primäre Hämochromatose (angeborene Formen)
Die erbliche (hereditäre) Hämochromatose ist die häufigste Form, sie wird gewöhnlich autosomal-rezessiv vererbt. Es gibt aber auch seltenere Gendefekte. Neben Gendefekten können auch andere Erkrankungen eine Eisenüberladung verursachen.
Je nach Manifestationsalter und zugrunde liegender Genveränderung kann unterschieden werden:
Klassische Form
- Typ 1: klassische Form, Synonyme: Symptomatische Form der klassischen Hämochromatose ; Symptomatische Form der HFE-Gen-assoziierten hereditären Hämochromatose, Mutationen im HFE-Gen (Chromosom 6 Genlocus p21.3) Genprodukt: Hereditäre-Hämochromatose-Protein, mit 90 % die häufigste Mutation[7]
Seltene Hämochromatoseformen
Seltene Formen umfassen:[8]
- Typ 2: juvenile Form, Synonyme: Jugendliche Form, Hämochromatose, juvenile[9]
- Typ 2A: Mutationen im HJV-Gen (Chromosom 1 Genlocus q21) Genprodukt: Hämojuvelin
- Typ 2B: Mutationen im HAMP-Gen (Chromosom 19 Genlocus q13.1) Genprodukt: Hepcidin
- Typ 3: Synonyme: Hämochromatose, TFR2-Gen-assoziierte, Mutationen im TFR2-Gen (Chromosom 7 Genlocus q22) Genprodukt: Transferrin-Rezeptor 2[10]
- Typ 4, Synonyme: Hämochromatose, autosomal-dominante ; Hämochromatose durch Ferroportin-Defekt ; Hämochromatose, hereditäre, autosomal-dominante, Mutationen im SLC40A1-Gen (Chromosom 2 Genlocus q32) Genprodukt: Ferroportin-1[11]
- Typ 5[12]
Neonatale Form
- Neonatale Form, sehr selten, tritt bereits im Kindes- oder Neugeborenenalter auf, ihr Verlauf ist meist sehr schwer.
Weitere angeborene Erkrankungen mit Eisenüberladung
- Acaeruloplasminämie
- Hereditäre Atransferrinämie und Hypotransferrinämie
- mit als wesentliches Merkmal beim GRACILE-Syndrom
Die Typen 1–3 werden jeweils autosomal-rezessiv vererbt und sind mit den typischen als Hämochromatose beschriebenen klinischen Symptomen vergesellschaftet. Dabei sind die Typen 1, 2A und 2B mit einer Erniedrigung des Hepcidin-Spiegels vergesellschaftet. Hepcidin hemmt durch Bindung an Ferroportin in Darm-Mucosazellen die Aufnahme von Eisen in den Blutkreislauf. Der Typ 1 mit der klassischen HFE-Mutation hat dabei ein geringeres Risiko für Organschäden als die Typen 2A und 2B.[13]
Die Typen 2A und 2B unterscheiden sich von den anderen Hämochromatoseformen durch ihren frühen Beginn von Symptomen und Organschäden. Sie treten bereits im zweiten bis dritten Lebensjahrzehnt auf und werden deshalb auch als juvenile (jugendliche) Hämochromatose bezeichnet. Der klassische Typ 1 und auch der Typ 4 manifestieren sich erst in der vierten bis fünften Lebensdekade.[13]
Der Typ 4 nimmt innerhalb der Hämochromatosen eine Sonderstellung ein. Er wird autosomal-dominant vererbt, hat ein geringeres Potential für Organschäden und ist erst im späteren Verlauf durch Laboruntersuchungen von Ferritin und Transferrin nachzuweisen. Manche Autoren schlagen daher den Typ 4 als eigene Krankheitsentität vor.[13]
Sekundäre Hämochromatose (erworbene Eisenüberladung)
Es sind mehrere verschiedene Ursachen für eine erworbene Eisenüberladung bekannt, zum Beispiel bei erhöhter Eisenzufuhr in den Körper. So können Menschen, die sehr viele Bluttransfusionen erhalten, eine sekundäre Hämochromatose entwickeln. Auch bei sehr starker Eisenaufnahme über den Magen-Darm-Trakt kann es zu einer erworbenen Eisenüberladung kommen. Diese Form ist vor allem im Afrika südlich der Sahara anzutreffen, da dort Spirituosen vorkommen, welche in Eisengefäßen gebrannt werden. Auch bei einer langdauernden Hämolyse kann eine Eisenüberladung entstehen, da der Körper sich mit dem aus den zerstörten roten Blutkörperchen gesammelten Eisen überlädt. Sehr selten sind genetische Ursachen, wie z. B. ein Mangel an Transferrin. Eine lange Hämodialysetherapie kann auch in manchen Fällen zu einer Hämochromatose führen.[14] Die Behandlung der sekundären Formen ist oft gleich wie die Behandlung der erblichen Hämochromatosen. Wenn aber gleichzeitig eine Anämie auftritt, können zur Therapie keine Aderlässe gemacht werden.[15]
Pathogenese
Der menschliche Körper verfügt über keinen Mechanismus zur aktiven Ausscheidung von Eisen. Ein „Abbau“ erfolgt nur bei Blutverlust. Allerdings kann eine weitere Aufnahme von Eisen verhindert werden, indem im Darm durch Hepcidin der Ferroportin-Transport von Eisen ins Blut gestoppt wird und das so in den Darm-Mucosazellen gesammelte Eisen mit den abgeschilferten Zellen wieder in das Darmlumen abgegeben wird. Durch Störung einzelner Komponenten des Eisenhaushalts kann diese Aufnahmehemmung verhindert werden und der Körper reichert folglich Eisen in verschiedenen Körperorganen, insbesondere der Leber, an.
Biochemische Schädigung
Der zu hohe Eisengehalt schädigt die Erbinformation der Zellen und sorgt auch für die Bildung schädlicher freier Radikale. Durch die ständigen Zellschädigungen verlieren die betroffenen Organe ihre Funktion. Infolgedessen entstehen Leberschäden und auch Leberkrebs. Hämochromatose kann neben den Leberschäden auch die Hirnareale für Geschlechtshormone schädigen oder eine Zuckerkrankheit verursachen. Auch ein Zusammenhang mit der Parkinson-Krankheit wurde vermutet.[16]
Genetische Ursachen
Verschiedene Mutationen von Genen, die den menschlichen Eisenhaushalt steuern, können eine Hämochromatose verursachen. Das HFE-Gen codiert für ein Protein, das mit dem Haupthistokompatibilitätskomplex der Klasse 1 strukturell verwandt ist. Im gesunden Menschen bildet das HFE-Protein HLA-H einen Komplex mit β2-Mikroglobulin und wird an der äußeren Seite der Zellmembran exprimiert. Der HLA-H-β2-Komplex ermöglicht die Bindung von Transferrin, des Haupteisentransportproteins im Blut. Der Komplex wird durch Endozytose aufgenommen. Das dabei entstandene Lysosom wird durch Ansäuerung aufgelöst und Eisen wird freigesetzt. Die Epithelzellen des Dünndarms, über die Eisen aufgenommen wird, steigern ihre Eisenaufnahme gegenläufig zum Eisenspiegel in ihrem Zellplasma. Daneben wird seit kurzem noch ein weiterer Regulationskreis über Hepcidin und Ferroportin diskutiert. Auch dieser ist bei der Hämochromatose Typ 1 betroffen,[3] da die HFE-Mutation auch mit einem Mangel an Hepcidin einhergeht. Der genaue Mechanismus ist bisher ungeklärt.[17]
Die Hämochromatose Typ 2A entsteht durch eine Mutation des Hämojuvelins und geht ebenso mit erniedrigten Hepcidinspiegeln einher. Hierbei fungiert Hämojuvelin als positiver Regulator der Hepcidintranskription, der sein Signal über den BMP-Protein-Signalweg weitergibt.[18][19]
Die Hämochromatose Typ 2B führt über eine Mutation des Gens für Hepcidin zu einem Hepcidinmangel. Dies führt zu einer verminderten Ausschleusung von Eisen aus den Darmzellen, Makrophagen und den Zellen der betroffenen Organe. Beim Typ 4 ist das Ferroportin – ein Protein, das direkt der Ausschleusung von Eisen aus den Zellen dient – defekt. Dies betrifft jedoch nur Leberzellen, Zellen der Plazenta und Makrophagen.[13]
Der gesunde menschliche Körper enthält rund 2–6 Gramm Eisen. Dieses ist zu 98 % in den Leberzellen (Hepatozyten) gespeichert. Die jährliche Eisenaufnahme beträgt beim Hämochromatosekranken rund 0,5–1,0 Gramm, variierend nach Geschlecht, Alkoholkonsum und Nahrungszusammensetzung. Ab einer kumulativen Eisenaufnahme von 20 Gramm entstehen die ersten Symptome. Der erste Haupteinlagerungsort sind die Leberzellen, die durch den erhöhten Eisengehalt durch mehrere Mechanismen geschädigt werden. Eisen selbst wirkt DNA-schädigend und kann über die Bildung von Radikalen Fette oxidieren. Daneben stimuliert Eisen über einen bislang noch unbekannten Mechanismus die Bildung von kollagenen Fasern im Extrazellularraum[14] und wird noch in der Bauchspeicheldrüse, dem Herzen und der Hirnanhangsdrüse gespeichert.[3]
Die Hämochromatose wird autosomal rezessiv vererbt. Die Erkrankung wird also in der Regel nur dann manifest, wenn beide Ausführungen des Gens den Defekt besitzen (homozygote Mutation). Schwächere Formen der Hämochromatose sind allerdings auch bei heterozygoten Mutationen möglich. Die Penetranz der Mutation ist gering, etwa 30 % der Männer mit homozygoter Mutation und nur etwa 1 % der homozygoten Frauen entwickeln ein klinisch relevantes Krankheitsbild; bei heterozygoten Merkmalsträgern ist das Auftreten einer Erkrankung sehr selten.[20]
Mutationen im HFE-Gen können auch krankhafte Veränderungen unabhängig von der Hämochromatose bedingen. Unter anderem wurden Erhöhungen der Bluttriglyceride beschrieben.[21]
Pathologie
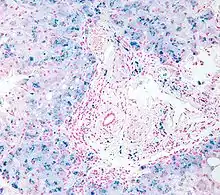
Eine Untersuchung von entnommenem Lebergewebe kann die Eisenüberladung der Leberzellen sichtbar machen. Sie gibt aber keinen Hinweis dahingehend, ob die Erkrankung genetische Ursachen hat oder durch eine andere Grunderkrankung verursacht wird.
Die krankhafte Eisenablagerung lässt sich lichtmikroskopisch feststellen. Die Ablagerungen zeigen sich in der HE-Färbung als grobe rostbraune Körnchen im Zellplasma. Die Ablagerungen beginnen typischerweise an den Leberzellen um die Portalfelder. Bei weiter fortschreitender Eisenspeicherung treten die Körnchen auch im Rest des Leberläppchens auf. Im späteren Verlauf zeigen auch Kupffer-Sternzellen sowie die Gallengangszellen entsprechende Auffälligkeiten. Sobald die Eisenspeicherung zu Gewebsschäden führt, werden Fibrose und Zirrhose der Leber sichtbar.[14] Die Eisenablagerungen lassen sich durch die Färbung mit Berliner Blau spezifisch nachweisen.[22]
Diagnose

Der Verdacht auf eine Hämochromatose sollte bei erhöhten Werten für Ferritin (> 200 μg/l bei Frauen, > 300 μg/l bei Männern) und Transferrinsättigung (> 45 % bei Frauen, > 50 % bei Männern) im Blut gestellt werden. In diesem Falle sollte ein Gentest auf das Vorliegen einer genetischen Hämochromatose erfolgen. Ist dieser negativ, sollte eine Leberbiopsie in Erwägung gezogen werden.[23][24]
Verschiedene Laborparameter können einen Hinweis auf eine Eisenüberladung liefern. So ist die Konzentration des Eisens selbst im Blutplasma i. d. R. erhöht. Die Eisenbindungskapazität ist dagegen i. d. R. bei einer Homozygotie erniedrigt oder im Normbereich. Bei heterozygot betroffenen Menschen ist sie dagegen manchmal erhöht, manchmal normwertig. Ebenso sind bei manifester Erkrankung die Transferrinsättigung und das Ferritin erhöht. Ferritin selbst ist bei symptomatisch Gesunden i. d. R. unter 500 μg/l. Bei symptomatisch Kranken kann sie bis auf 6.000 μg/l erhöht sein. Zur Unterscheidung gegenüber einer fortgeschrittenen Lebererkrankung durch Alkoholkonsum kann der Ferritinspiegel einen Hinweis geben, da bei alkoholischer Lebererkrankung der Ferritinspiegel unter 500 μg/l liegen sollte. Die Laborveränderungen sind aber nicht vollkommen spezifisch, da auch andere Lebererkrankungen den Eisenspiegel in der Leber erhöhen können.[3] Ferritin ist ein Akute-Phase-Protein und infolgedessen bei entzündlichen Prozessen generell erhöht.[5] Die Transferrinsättigung ist dabei der sensitivste Laborparameter zum Nachweis einer Hämochromatose im asymptomatischen Stadium.[25] Die endgültige Diagnose sollte durch eine genetische Untersuchung des HFE-Gens gestellt werden. Sollte der Gentest negativ sein, kann die Diagnose durch eine feingewebliche Untersuchung (Leberbiopsie, Histologie mit Berliner-Blau-Färbung) gestellt werden.[26]
Als weitere diagnostische Methode ist die quantitative Bestimmung des Eisengehalts aus unfixiertem Lebergewebe möglich. Der Normalwert liegt dabei unter 1.000 μg/g Trockenmasse. Menschen mit erblicher Hämochromatose weisen Werte über 10.000 μg/g auf. Mit irreversiblen Leberschäden und Zirrhose ist ab 22.000 μg/g zu rechnen,[14] ist aber aufgrund der geringen Penetranz der Erkrankung nur in Zusammenschau mit den klinischen und laborchemischen Befunden aussagekräftig.[5]
Der Eisengehalt der Leber kann auch nichtinvasiv durch eine Computertomographie oder eine Magnetresonanztomographie bestimmt werden. Diese Methoden sind aber nur semiquantitativ und somit nur begrenzt aussagekräftig.[5] Die Etablierung von Hepcidin als diagnostischer Test für Hämochromatose wird derzeit diskutiert.[27][28]
Früherkennung
In Fachkreisen wurde die Hämochromatose als mögliche Krankheit benannt, bei der eine allgemeine Untersuchung der Gesamtbevölkerung sinnvoll wäre. Jüngere Studien ziehen dies jedoch mittlerweile in Zweifel.
Die Bundesärztekammer identifizierte 2003 die Hämochromatose als eine Krankheit, bei der nach ihrer Meinung ein generelles Screening der Bevölkerung vorteilhaft wäre.[29] Eine Genotypisierung wird mittlerweile ab einer Transferrinsättigung von 45 % in zwei verschiedenen Tests empfohlen.[30] Andere Veröffentlichungen empfehlen einen Cutoff von 55 %.[31] 2006 kam die von der US-Regierung eingerichtete Preventive Services Task Force zu dem Ergebnis, dass die genetischen Grundlagen der Hämochromatose zu wenig erforscht seien, als dass ein generelles Screening empfohlen werden könne.[32] Eine kanadische Forschergruppe sprach sich 2009, nach einem Testscreening an 100.000 Menschen, gegen ein allgemeines Screening aus. Außerdem kam sie zu dem Schluss, dass die Transferrinsättigung oder der Ferritinspiegel ungenügend spezifisch wären und somit für ein Screening nicht geeignet seien.[33]
Therapie
Physikalische Verfahren
Die Therapie der Wahl ist der Aderlass und besteht aus zwei Phasen, die sich üblicherweise wie folgt gestalten:[34][35]
- Phase 1 (Initialtherapie): wöchentliche Aderlässe von 500 ml, bis der Ferritinspiegel unter 50 μg/l gefallen ist;
- Phase 2 (Langzeittherapie): lebenslange Aderlässe von 500 ml, um den Ferritinspiegel im weiteren Verlauf zwischen 50 und 100 μg/l zu halten (etwa 4–12 pro Jahr).
Ziel der Therapie ist eine Entleerung oder zumindest Reduzierung der Eisenspeicher, was am wirksamsten durch eine Aderlasstherapie erreicht wird. Anfangs sollte ein Aderlass von 500 ml einmal bis zweimal pro Woche durchgeführt werden. Ein halber Liter Blut enthält rund 200–250 mg Eisen. Es sollte eine wöchentliche Behandlung durchgeführt werden, bis die Serumferritinspiegel unter 50 μg/l fallen. Dies kann je nach Alter und Eisenbeladung bis zu mehreren Jahren dauern. Zur Erhaltungstherapie können dreimonatliche Aderlässe durchgeführt werden. Diese sind in der Regel zur Aufrechterhaltung eines Plasmaferritinspiegels von 50 bis 100 μg/l ausreichend, der das Langzeittherapieziel darstellt.[3] Andere Angaben empfehlen auch einen Zielspiegel unter 50 μg/l.[5] Dabei ist zu beachten, dass die Aderlässe möglichst regelmäßig durchgeführt werden, damit sich eine konstante Regeneration der verlorenen Blutmenge einstellt.[3] Eine Eisenmangelanämie tritt charakteristischerweise bei den Typen 1 bis 3 nicht auf. Erkrankte mit dem Typ 4 sollten engmaschig überwacht werden, da bei ihnen Anämien häufiger auftreten. Ebenso hat bei ihnen die Aderlasstherapie einen geringeren Effekt.[13]
Eine andere Therapieform ist die Erythroapherese, wobei mehr Erythrozyten pro Behandlung entnommen werden können, wodurch die Häufigkeit der Behandlung reduziert werden kann und der Ferritinwert schneller absinkt als bei der einfachen Aderlasstherapie. Diese Therapieform ist allerdings aufwendiger, die Kostenübernahme durch die Krankenkassen noch nicht geklärt.
Medikamente
Protonenpumpenhemmer haben einen hemmenden Effekt auf die Resorption von nicht-haem-gebundenem Eisen und können die notwendige Menge und Frequenz der Aderlässe vermindern.[36]
Auch die Gabe von Deferoxamin dient der Eisenreduktion, diese Therapie ist aber nicht so wirksam. Sie wird nur angewandt, wenn eine Blutarmut (Anämie) oder fortgeschrittene Herzmuskelschwäche (Kardiomyopathie) bestehen. Die Deferoxaminbehandlung ist aufwändig (Dauerinfusion an 5–7 Tagen pro Woche), hat häufig Nebenwirkungen (Seh- und Hörstörungen) und ist weniger wirksam als der Aderlass oder die Apherese. Mittlerweile ist auch ein Präparat zum Schlucken (Deferasirox) verfügbar.[5][15] Falls aufgrund einer bereits fortgeschrittenen Herzinsuffizienz eine Aderlasstherapie nicht vertretbar scheint, müssen Eisenbinder eingesetzt werden. Dabei sollten die beiden verschiedenen Wirkstoffe kombiniert zugeführt werden. Da sie sich in ihrer Diffusionsstärke in das Zellinnere unterscheiden, können die Eisenspeicher schneller entleert werden.[37] Eine weitere Substanz zur Eisenreduktion ist Deferipron (Handelsname Ferriprox). Es wird schnell resorbiert und erreicht nach 45 Minuten den höchsten Serumspiegel. 85 % werden durch Glukuronisierung (Verbindung mit Glukuronsäure) harnfähig gemacht. Das Glukuronid des Deferiprons bindet Eisen und scheidet dieses mit dem Urin aus. Mögliche Nebenwirkungen sind Übelkeit, Bauchschmerzen, Erbrechen, erhöhte Leberwerte, Gelenkschmerzen und Neutropenie (Verminderung weißer Blutkörperchen).[38]
Die Symptome der Gelenkmanifestation der Erkrankung können mit NSAR abgemildert werden. Ebenso profitieren viele Patienten von einer Osteoporosebehandlung.[39]
Diätetische Maßnahmen
Diätetische Maßnahmen können die Heilung unterstützen. Konkret sollten stark eisenhaltige Nahrungsmittel zurückhaltend konsumiert werden. Schwarzer Tee oder auch Milch, zusammen mit der Mahlzeit getrunken, vermindert die Eisenabsorption. Umgekehrt sollte auf den Konsum Vitamin-C-haltiger Getränke (z. B. Orangensaft) im Zeitraum von etwa zwei Stunden vor bis zwei Stunden nach den Mahlzeiten verzichtet werden, da Vitamin C die Aufnahme von Eisen aus der Nahrung begünstigt. Da der Konsum von Alkohol die Eisenaufnahme steigert, ist Alkoholkarenz sinnvoll.[5]
Organtransplantation
Bei fortgeschrittenem Leberschaden kann im Falle eines Leberversagens eine Lebertransplantation durchgeführt werden. Dies ist in manchen Fällen auch möglich, wenn die Patienten ein Leberzellkarzinom entwickelt haben. Transplantierte Hämochromatose-Patienten haben aber aufgrund der meist vorhandenen, durch ihre Grunderkrankung hervorgerufenen, Begleiterkrankungen eine schlechtere Prognose als Transplantierte mit anderer Grunderkrankung.[26]
Prognose
„Die Hämochromatose kann durch die Schädigung der Leber zu lebensbedrohlichen Situationen führen. Ebenso erhöht sie stark das Risiko für Leberzellkrebs, falls sie nicht vor dem Beginn der Symptome entdeckt wird. Wird die Krankheit vor Auftreten der Symptome entdeckt und behandelt, gilt sie als ohne Folgeschäden heilbar. Ohne Therapie ist die Prognose hingegen infaust.“[34]
Wird die Erkrankung vor dem Auftreten irreversibler Organveränderungen behandelt, so wirkt sie sich nicht nachteilig auf die Lebenserwartung aus. Leberfibrose und Leberzirrhose sind nicht mehr rückgängig zu machen und erfordern eine eigenständige Behandlung.[3] Die Wahrscheinlichkeit des Auftretens von Langzeitkomplikationen, einschließlich des Leberzellkarzinoms, steigt mit der Dauer und dem Ausmaß der Eisenüberladung. Infolgedessen ist eine frühe Diagnosestellung entscheidend.[40] Bereits bei Diagnosestellung bestehende strukturelle Schäden an den Gelenken[39] und Geschlechtsorganen werden als irreversibel betrachtet. Das Fortschreiten der Veränderungen kann durch eine Therapie jedoch verlangsamt werden.[13] Rund 35 % der an einer manifesten Hämochromatose erkrankten Menschen entwickeln im späteren Verlauf ein Leberzellkarzinom.[41]
Medizingeschichte

1865 beschrieb Armand Trousseau ein klinisches Syndrom, bestehend aus Leberzirrhose, Diabetes und bronzefarbener Hautpigmentierung.
Aufgrund weiterer derartiger Beschreibungen aus dem Jahre 1871 durch Charles Émile Troisier und 1882 durch Victor Charles Hanot und Anatole Chauffard wurde die historische Bezeichnung Troisier-Hanot-Chauffard-Syndrom gebräuchlich.[42]
1889 prägte Friedrich Daniel von Recklinghausen den Begriff Hämochromatose. 1935 erkannte Joseph H. Sheldon die erbliche Komponente der Erkrankung.[25] Bis dahin war die Hämochromatose fälschlicherweise auf Alkoholmissbrauch zurückgeführt worden.[43] In den 1970er-Jahren wurde der autosomal-rezessive Erbgang der Typen 1 bis 3 erkannt.[13] Eine US-amerikanische Forschungsgruppe sequenzierte 1996 das HFE-Gen und stellte dessen Verbindung zur Hämochromatose dar.[25] Es wird vermutet, dass die HFE C282Y-Mutation vor etwa 4000 Jahren bei einem Menschen in Mitteleuropa, der vermutlich keltischer Abstammung gewesen ist, aufgetreten ist und sich, von dort ausgehend, mit dessen Nachkommen in der europäischen Population ausgebreitet hat.[44] Eine mögliche Hypothese für die Verbreitung der Mutation ist die Vermutung, dass die übermäßige Eisenspeicherung bei längerer Unterversorgung mit Eisen einen Überlebensvorteil bieten könne.[13]
Hämochromatose bei Tieren
Die Hämochromatose ist bei Tieren sehr selten.[45] So wurden bei Hunden Einzelfälle von sekundären Hämochromatosen bei Pyruvatkinase-Mangel[46] und nach wiederholten Bluttransfusionen[47] beobachtet. Bedlington Terrier weisen indes genetische Prädispositionen auf. Bei Rindern gab es einige Hämochromatose-Fälle bei Tieren der französischen Rinderrasse Salers und ihren Kreuzungen.[45] Bei Vögeln kommen Eisenüberladungen mit Hämochromatose-ähnlichem klinischen Bild vor allem bei Staren vor.[48] Auch bei Pferden ist ein Fall von Eisenüberladung mit Leberschäden beschrieben.[49] Darüber hinaus existieren einige Tiermodelle mit genetisch-veränderten Labornagern sowie durch exzessive Eisenzufuhr.
Weblinks
- P. C. Adams, J. C. Barton: How I treat hemochromatosis. In: American Society of Hematology (Hrsg.): Blood. Band 116, Nr. 3, 22. Juli 2010, S. 317–325, doi:10.1182/blood-2010-01-261875, PMID 20308595 (englisch, bloodjournal.org): “standard therapy is the weekly removal of blood, authors declare no competing financial interests”
- HFE. In: Online Mendelian Inheritance in Man. (englisch)
- HJV. In: Online Mendelian Inheritance in Man. (englisch)
- Neonatale Hämochromatose. In: Online Mendelian Inheritance in Man. (englisch)
- Hämochromatose Vereinigung Deutschland e.V.
- Bild der Hautveränderungen bei Hämochromatose. In: R. Lim u. a.: A permanent tan from iron. Kidney Int. 2008 Apr;73(7), S. 898. PMID 18340355.
Literatur
Lehrbücher
- Lawrie W. Powell: Hemochromatosis. In: Anthony S. Fauci u. a.: Harrison's Principles of Internal Medicine. 17. Auflage. New York 2008, S. 2429–2433.
- Gerd Herold u. a.: Innere Medizin. Eine vorlesungsorientierte Darstellung ; unter Berücksichtigung des Gegenstandskataloges für die Ärztliche Prüfung ; mit ICD 10-Schlüssel im Text und Stichwortverzeichnis. Herold, Köln 2013, ISBN 978-3-9814660-2-7, S. 543.
Artikel in Fachzeitschriften
- A. Pietrangelo: Hemochromatosis: an endocrine liver disease. In: Hepatology. 2007 Oct;46(4), S. 1291–1301. PMID 17886335.
- Robson, Merryweather-Clarke, Pointon u. a.: Diagnosis and management of haemochromatosis since the discovery of the HFE gene: a European experience. In: Br J Haematol. 2000 Jan;108(1), S. 31–39. PMID 10651721.
- W. Griffiths, T. Cox: Haemochromatosis: novel gene discovery and the molecular pathophysiology of iron metabolism. In: Hum. Mol. Genet. Band 9, Nr. 16, Oktober 2000, S. 2377–2382, PMID 11005792 (hmg.oxfordjournals.org).
- K. Pantopoulos: Function of the hemochromatosis protein HFE: Lessons from animal models. In: World J. Gastroenterol. Band 14, Nr. 45, Dezember 2008, S. 6893–6901, doi:10.3748/wjg.14.6893, PMID 19058322, PMC 2773850 (freier Volltext) – (wjgnet.com).
Einzelnachweise
- Uniklinik Ulm – Hämochromatose-Ambulanz – Definition der Hämochromatose (Memento vom 27. Mai 2009 im Internet Archive) (Seite abgerufen am 6. Dezember 2009)
- C. Y. Lok, A. T. Merryweather-Clarke, u. a.: Iron overload in the Asian community. In: Blood. Band 114, Nr. 1, 2. Juli 2009, S. 20–25, doi:10.1182/blood-2009-01-199109, PMID 19342478.
- Lawrie W. Powell: Hemochromatosis. In: Anthony S. Fauci u. a.: Harrison's Principles of Internal Medicine. 17. Auflage. New York 2008, S. 2429–2433.
- Robson, Merryweather-Clarke, Pointon u. a.: Diagnosis and management of haemochromatosis since the discovery of the HFE gene: a European experience. In: British Journal of Haematology. Band 108, Nr. 1, Januar 2000, S. 31–39, PMID 10651721.
- Gerd Herold u. a.: Innere Medizin. Köln 2013, S. 543–545.
- Carlos Thomas (Hrsg.): Spezielle Pathologie. Schattauer Verlag, 1996, ISBN 3-7945-1713-X, S. 379 (books.google.at).
- Hämochromatose Typ 1, symptomatische Form. In: Orphanet (Datenbank für seltene Krankheiten).
- Hämochromatose, hereditäre seltene. In: Orphanet (Datenbank für seltene Krankheiten).
- Hämochromatose Typ 2. In: Orphanet (Datenbank für seltene Krankheiten).
- Hämochromatose Typ 3. In: Orphanet (Datenbank für seltene Krankheiten).
- Hämochromatose Typ 4. In: Orphanet (Datenbank für seltene Krankheiten).
- Hämochromatose Typ 5. In: Orphanet (Datenbank für seltene Krankheiten).
- A. Pietrangelo: Hereditary hemochromatosis--a new look at an old disease. In: The New England Journal of Medicine. Band 350, Nr. 23, 3. Juni 2004, S. 2383–2397, doi:10.1056/NEJMra031573, PMID 15175440.
- Chen Liu, James M. Crawford: The Gastrointestinal Tract. In: Vinay Kumar, Abul K. Abbas, Nelson Fausto: Robbins and Cotran – Pathologic Basis of Disease. 7. Auflage. Philadelphia 2005, S. 908–911.
- N. Gattermann: The treatment of secondary hemochromatosis. In: Dtsch Arztebl Int. 2009 Jul; 106 (30), S. 499–504, PMID 19727383.
- P. G. Mastroberardino, E. K. Hoffman, M. P. Horowitz u. a.: A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. In: Neurobiol. Dis. Band 34, Nr. 3, Juni 2009, S. 417–431, doi:10.1016/j.nbd.2009.02.009, PMID 19250966, PMC 2784936 (freier Volltext).
- S. Vaulont, D. Q. Lou, L. Viatte, A. Kahn: Of mice and men: the iron age. In: J. Clin. Invest. Band 115, Nr. 8, August 2005, S. 2079–2082, doi:10.1172/JCI25642, PMID 16075054, PMC 1180554 (freier Volltext).
- J. L. Babitt, F. W. Huang, D. M. Wrighting u. a.: Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. In: Nat. Genet. Band 38, Nr. 5, Mai 2006, S. 531–539, doi:10.1038/ng1777, PMID 16604073 (upenn.edu [PDF]).
- P. B. Yu, C. C. Hong, C. Sachidanandan u. a.: Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. In: Nat. Chem. Biol. Band 4, Nr. 1, Januar 2008, S. 33–41, doi:10.1038/nchembio.2007.54, PMID 18026094, PMC 2727650 (freier Volltext).
- Katrina J. Allen u. a.: Iron-Overload-Related Disease in HFE Hereditary Hemochromatosis. In: N Engl J Med. Nr. 358, 2008, S. 221–230.
- M. Solanas-Barca, R. Mateo-Gallego u. a.: Mutations in HFE causing hemochromatosis are associated with primary hypertriglyceridemia. In: J Clin Endocrinol Metab. 2009 Nov;94(11), S. 4391–4397 PMID 19820015.
- C. Thomas: Histopathologie. Stuttgart 1998, S. 165.
- Uniklinik Ulm – Hämochromatose-Ambulanz (Memento vom 12. März 2010 im Internet Archive) (Seite abgerufen am 2. Dezember 2009)
- Hämochromatose-Vereinigung Deutschland e. V. – Diagnose der Hämochromatose (Seite abgerufen am 2. Dezember 2009)
- A. Pietralango: Haemochromatosis. In: Gut. 52 Suppl 2, Mai 2003, S. ii23–ii30, doi:10.1136/gut.52.suppl_2.ii23, PMID 12651879, PMC 1867747 (freier Volltext).
- C. A. Whittington, K. V. Kowdley: Review article: haemochromatosis. In: Alimentary Pharmacology & Therapeutics. Band 16, Nr. 12, Dezember 2002, S. 1963–1975, PMID 12452931.
- A. Floreani, F. Navaglia, E. R. Rizzotto, D. Basso, M. Chiaramonte: Mass spectrometry measurement of plasma hepcidin for the prediction of iron overload. In: Clinical Chemistry and Laboratory Medicine. Band 49, Nr. 2, Februar 2011, S. 197–206, doi:10.1515/CCLM.2011.055, PMID 21143008.
- E. H. Kemna, H. Tjalsma, H. L. Willems, D. W. Swinkels: Hepcidin: from discovery to differential diagnosis. In: Haematologica. Band 93, Nr. 1, Januar 2008, S. 90–97, doi:10.3324/haematol.11705, PMID 18166790.
- Bekanntmachung der Bundesärztekammer: Richtlinien zur prädiktiven genetischen Diagnostik. In: Deutsches Ärzteblatt. Heft 6, Juni 2003, online abrufbar als PDF-Datei (Memento vom 26. Januar 2005 im Internet Archive).
- J. Zlocha, L. Kovács u. a.: [Molecular genetic diagnostics and screening of hereditary hemochromatosis]. In: Vnitrni Lekarstvi. Band 52, Nr. 6, Juni 2006, S. 602–608, PMID 16871764.
- C. E. Wrede, S. Hutzler, L. C. Bollheimer, R. Buettner, C. Hellerbrand, J. Schöelmerich, K. D. Palitzsch: Correlation between iron status and genetic hemochromatosis (codon C282Y) in a large German population. In: The Israel Medical Association journal : IMAJ. Band 6, Nummer 1, Januar 2004, S. 30–33, PMID 14740507.
- E. P. Whitlock, B. A. Garlitz, E. L. Harris, T. L. Beil, P. R. Smith: Screening for hereditary hemochromatosis: a systematic review for the U.S. Preventive Services Task Force. In: Annals of Internal Medicine. Band 145, Nr. 3, 1. August 2006, S. 209–223, PMID 16880463.
- P. Adams, J. C. Barton, G. D. McLaren u. a.: Screening for iron overload: lessons from the hemochromatosis and iron overload screening (HEIRS) study. In: Canadian Journal of Gastroenterology = Journal Canadien De Gastroenterologie. Band 23, Nr. 11, November 2009, S. 769–772, PMID 19893773, PMC 2777090 (freier Volltext).
- Uniklinik Ulm – Hämochromatose-Ambulanz – Therapie der Hämochromatose (Memento vom 18. Mai 2015 im Internet Archive) (Seite abgerufen am 22. Dezember 2009)
- Hämochromatose-Vereinigung Deutschland e. V. – Therapie der Hämochromatose (Seite abgerufen am 6. Dezember 2009)
- C. Hutchinson, C. A. Geissler, J. J. Powell, A. Bomford: Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. In: Gut. 2007;56, S. 1291–1295, PMID 17344278.
- W. Gilles, W. Stremmel: Kardiale Hämochromatose. In: Kardiologe. 2009, 3, S. 57–66. doi:10.1007/s12181-008-0125-6.
- Robert C. Hider, A. Victor Hoffbrand: The Role of Deferiprone in Iron Chelation. In: New England Journal of Medicine. Band 379, Nr. 22, 29. November 2018, ISSN 0028-4793, S. 2140–2150, doi:10.1056/NEJMra1800219 (nejm.org [abgerufen am 15. Februar 2019]).
- B. Möller: Hämochromatose-assoziierte Arthropathie – moderne Diagnostik für eine altmodische Therapie? In: Akt Rheumatol. 2008; 33, S. 281–289. doi:10.1055/s-2008-102763.
- C. Niederau, R. Fischer, A. Pürschel: Long-term survival in patients with hereditary hemochromatosis. In: Gastroenterology. 1996 Apr;110(4), S. 1107–1119. PMID 8613000.
- H. Spangenberg, R. Thimme, H. Blum: Der Leberrundherd. In: Deutsches Ärzteblatt. Jg. 104, Heft 33, August 2007, S. 17. (aerzteblatt.lnsdata.de (Memento vom 31. März 2010 im Internet Archive) PDF).
- Who named it
- A. Pietrangelo: Hemochromatosis: an endocrine liver disease. In: Hepatology. 2007 Oct;46(4), S. 1291–1301. PMID 17886335.
- S. Distante, K. J. Robson, J. Graham-Campbell, A. Arnaiz-Villena, P. Brissot, M. Worwood. The origin and spread of the HFE-C282Y haemochromatosis mutation. Hum Genet. 2004 Sep;115(4), S. 269–279. PMID 15290237
- D. O’Toole u. a.: Hepatic failure and hemochromatosis of Salers and Salers-cross cattle. In: Veterinary Pathology. Band 38, Nr. 4, Juli 2001, S. 372–389, doi:10.1354/vp.38-4-372, PMID 11467471 (vetpathology.org – Volltext). vetpathology.org (Memento vom 21. November 2008 im Internet Archive)
- J. W. Harvey: Pathogenesis, laboratory diagnosis, and clinical implications of erythrocyte enzyme deficiencies in dogs, cats, and horses. In: Vet Clin Pathol. 35 (2006 ), S. 144–156. PMID 16783707
- W. S. Sprague u. a.: Hemochromatosis secondary to repeated blood transfusions in a dog. In: Veterinary Pathology. Band 40, Nr. 3, Mai 2003, S. 334–337, doi:10.1354/vp.40-3-334, PMID 12724577.
- S. J. Gosselin, L. W. Kramer: Pathophysiology of excessive iron storage in mynah birds. In: J Am Vet Med Assoc. 183 (1983), S. 1238–1240.
- J. P. Lavoie, E. Teuscher: Massive iron overload and liver fibrosis resembling haemochromatosis in a racing pony. In: Equine Veterinary Journal. Band 25, Nr. 6, November 1993, S. 552–554, PMID 8276008.