Common Technical Document
Das Common Technical Document (CTD) ist ein vorgeschriebenes Dokumentenformat, in dem ein Pharmaunternehmen die pharmazeutische Qualität, Unbedenklichkeit und Wirksamkeit eines Arzneimittels im Rahmen der Arzneimittelzulassung dokumentieren und bei den Arzneimittelbehörden einreichen muss.
Geschichte und Bedeutung
Das Common Technical Document wurde im Rahmen des International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) zur Harmonisierung der Arzneimittelzulassungsbedingungen in der Europäischen Union, den USA und Japan entwickelt. Eine erste Version der Spezifikation wurde 2000 verabschiedet; zurzeit ist die dritte Revision von Januar 2004 gültig. Das Common Technical Document ist inzwischen in allen drei Regionen für die meisten Einreichungen vorgeschrieben; es wurde außerdem von Kanada und der Schweiz übernommen. Durch die Harmonisierung ist es möglich, in verschiedenen Ländern für ein neues Arzneimittel weitgehend identische Zulassungsunterlagen einzureichen.
Struktur
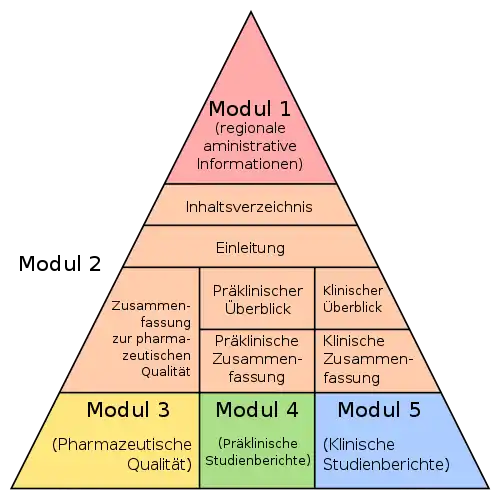
Die Struktur eines CTD besteht aus fünf Modulen:
- Modul 1: Administrative Information
- Modul 2: Inhaltsverzeichnis, Überblick und Zusammenfassungen der Module 3–5
- Modul 3: Pharmazeutische Qualität (Nachweis der Qualität)
- Modul 4: Präklinische Studienberichte (Nachweis der Wirksamkeit und Unbedenklichkeit)
- Modul 5: Klinische Studienberichte (Nachweis der Wirksamkeit und Unbedenklichkeit)
Modul 1
Das Modul 1 enthält im Gegensatz zu den anderen Modulen von Region zu Region unterschiedliche Dokumente. Deshalb wird das Modul 1 nicht als Teil des CTD angesehen. In Europa sind folgende Elemente im Modul 1 zu finden:
- 1.1 Umfassendes Inhaltsverzeichnis des gesamten Dossiers
- 1.2 Antragsformular zur Zulassung
- 1.3 Produktinformation: Zusammenfassung der Merkmale des Arzneimittels (SmPC), Packungsbeilage, Informationen zur Verpackung
- 1.4 Information über beteiligte Experten
- 1.5 Spezifische Anforderungen für verschiedene Antragsarten
- 1.6 Beurteilung von Umweltrisiken
- 1.7 Information zur Marktexklusivität für Orphan-Arzneimittel
- 1.8 Information zum Pharmakovigilanz-System
- 1.9 Information zu außerhalb der EU durchgeführten klinischen Studien
Modul 2
Modul 2 gibt einen Überblick über die im Dossier vorhandene Information zum Arzneimittel und integriert diese mit Beurteilungen und Schlussfolgerungen.
- 2.1 Inhaltsverzeichnis der Module 2 bis 5
- 2.2 Einleitung (eine Seite): kurze Darstellung der pharmazeutischen Klasse, des Wirkungsmechanismus und der beabsichtigten klinischen Anwendung.
- 2.3 Zusammenfassung zur pharmazeutischen Qualität (nicht mehr als 40 Seiten; für biotechnologische Pharmazeutika: 80 Seiten): Überblick und Zusammenfassung der Daten aus Modul 3.
- 2.4 Nichtklinischer Überblick (nicht mehr als 30 Seiten): integrierte Analyse der nichtklinischen Information im Dossier (Übersichten zur nichtklinischen Teststrategie, Pharmakologie, Pharmakokinetik und Toxikologie sowie ein integrierender Überblick und Schlussfolgerungen). Es muss angegeben werden, inwieweit die Studien in Übereinstimmung mit der Good Laboratory Practice durchgeführt wurden.
- 2.5 Klinischer Überblick (zirka 30 Seiten): integrierte Analyse der klinischen Information im Dossier (Begründung zur Produktentwicklung, Übersichten zur Biopharmazie, klinischen Pharmakologie, Wirksamkeit, Sicherheit sowie Schlussfolgerungen zu Nutzen und Risiken). Es muss angegeben werden, inwieweit die Studien in Übereinstimmung mit der Good Clinical Practice durchgeführt wurden.
- 2.6 Nichtklinische Zusammenfassungen und Tabellenübersichten über alle durchgeführten, in Modul 4 dargestellten nichtklinischen Studien zur Pharmakologie, Pharmakokinetik und Toxikologie (nicht mehr als 100–150 Seiten).
- 2.7 Klinische Zusammenfassungen zu biopharmazeutischen Studien, klinischen pharmakologischen Studien, klinischen Studien zur Wirksamkeit, Zusammenfassungen zur klinischen Sicherheit, Literaturangaben sowie Synopsen der individuellen, in Modul 5 dargestellten klinischen Studien (üblicherweise 50–400 Seiten).
Modul 3
Modul 3 stellt ausführlich die chemisch-pharmazeutischen und biologischen Daten zum Arzneistoff und zum als Arzneiform vorliegenden Produkt dar. Alternativ kann zu Inhaltsstoffen eine Konformitätsbescheinigung (CEP) der EDQM vorgelegt werden; eine detaillierte Darstellung zur Herstellung und Qualitätskontrolle im Modul 3 entfällt dann für diese Stoffe. Für Daten zu Arzneistoffen kann auch auf ein Drug Master File verwiesen werden, wenn der Arzneistoffhersteller und der Arzneimittelhersteller nicht identisch sind.
- 3.1 Inhaltsverzeichnis für Modul 3
- 3.2.S Daten zum Stoff, der Struktur, den Stoffeigenschaften, dem Hersteller, dem Herstellungsprozess, der Inprozesskontrolle, den Ausgangsmaterialien, den Zwischenprodukten, der Prozessvalidierung sowie zur Entwicklung des Herstellungsprozesses, ferner Information zur Stoffcharakterisierung, zu Verunreinigungen, Spezifikationen, Analysemethoden und deren Validierung, zu Referenzsubstanzen und schließlich Daten zur Stabilität des Stoffes unter verschiedenen Umweltbedingungen.
- 3.2.P Daten zum Produkt; Beschreibung und genaue Zusammensetzung des Arzneimittels für alle Arzneiformen und Dosierungen, darunter Informationen zu allen Hilfsstoffen sowie wiederum Daten zu Herstellung, Prozessvalidierung, Analyse, Primärpackmittel und Stabilität des fertigen Produktes.
- 3.2.A Anhänge, die gegebenenfalls die Beschreibung besonderer Prozessanlagen oder Ausrüstungen, die Sicherheitsbeurteilung von Zusatzstoffen oder die Beschreibung neuartiger Hilfsstoffe enthalten.
- 3.2.R Regionale Informationen, bei denen es sich um für die entsprechende Region (Europäische Union, USA oder Japan) spezifische Dokumente zur Qualität des Arzneimittels oder des Wirkstoffs handelt.
- 3.3 Literaturangaben
Modul 4
Modul 4 stellt ausführlich sämtliche Studienberichte zu allen nichtklinischen (= nicht beim Menschen durchgeführten) Studien dar.
- 4.1 Inhaltsverzeichnis für Modul 4
- 4.2 Studienberichte zur Pharmakologie (Pharmakodynamik, Sicherheitspharmakologie, Pharmakodynamische Arzneimittelwechselwirkungen), Pharmakokinetik und Toxikologie (Toxizitätsbestimmungen, Mutagenitätsprüfung, Karzinogenitätstests, Reproduktionstoxikologie, lokale Verträglichkeit, Immuntoxikologie und weitere)
- 4.3 Literaturangaben
Modul 5
Modul 5 stellt ausführlich sämtliche Studienberichte zu allen klinischen (= beim Menschen durchgeführten) Studien dar.
- 5.1 Inhaltsverzeichnis für Modul 5
- 5.2 Tabellarische Übersicht über sämtliche klinische Studien
- 5.3 Studienberichte zur Biopharmazie, Pharmakokinetik, Pharmakodynamik, Wirksamkeit und Sicherheit, ferner Informationen zu Erfahrungen mit dem schon vermarkteten Produkt (so vorhanden) und sämtliche Case Report Forms und eine Auflistung aller teilnehmenden Patienten.
- 5.4 Literaturangaben
Elektronische Einreichung
Das Common Technical Document definiert ein Papierformat; ein vollständiges Dossier umfasst oft mehrere zehntausend Seiten. Um diese Datenmengen für Antragsteller und Behörden besser handhabbar zu machen, wurde im Rahmen der ICH ein elektronisches Format, das eCTD, definiert, welches inhaltlich auf dem CTD basiert.
Weitere Verwendungen des CTD-Formates
Über den Zulassungsantrag für ein neues Arzneimittel hinaus wird das CTD-Format inzwischen auch zur Pflege der Zulassungsunterlagen genutzt. Pharmazeutische Unternehmen müssen Änderungen bei Zulassungsdaten, sogenannte Änderungsanzeigen, im CTD-Format einreichen.
Auch das Drug Master File, das zum Einsatz kommt, wenn der Hersteller des Arzneistoffes und der Hersteller des Arzneimittels nicht identisch sind, verwendet das Format des CTD Teil 3.2.S.
Schließlich werden Teile des CTD-Formates bei Genehmigungsanträgen für klinische Studien genutzt. In diesen Anträgen muss ein Dossier über das Prüfpräparat, in Europa ein sogenanntes Investigational Medicinal Product Dossier (IMPD), eingereicht werden, das in der Struktur dem Modul 3 des CTD folgt.
Quellen
- The Common Technical Document, ICH M4
- Volume 2B: Notice to Applicants - Medicinal products for human use (PDF-Datei; 1,01 MB)