SELEX
Unter dem Akronym SELEX (engl.: Systematic Evolution of Ligands by EXponential Enrichment, zu deutsch: Systematische Evolution von Liganden durch exponentielle Anreicherung) versteht man in der Molekularbiologie ein kombinatorisches Verfahren zur gerichteten Evolution von Oligonukleotid-Strängen, beispielsweise einsträngiger DNA oder RNA. Diese können als Liganden spezifisch an ausgewählte Targets binden.[1][2]
Eigenschaften
Die mit Hilfe der SELEX gewonnenen Oligonukleotid-Sequenzen werden Aptamere genannt. Das Verfahren wird gelegentlich auch als In-vitro-Selektion oder In-vitro-Evolution bezeichnet.
Einzelstrang-DNA-basierte (ssDNA) Aptamere sind nach Ellington und Szostak[3] wegen ihrer höheren Stabilität bei gleicher Spezifität besser geeignet als RNA-basierte Aptamere.[4]
SELEX® ist ein eingetragenes Warenzeichen der Firma Gilead Sciences.
Das Verfahren
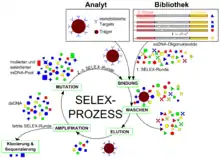
Zu Beginn des Verfahrens besteht die Synthese aus einer sehr großen Anzahl von unterschiedlichen Oligonukleotiden, die so genannte Molekülbibliothek, auch Molekülpool genannt. Aus diesem Pool von bis zu 1016 und mehr unterschiedlichen Molekülen werden die Moleküle mit den gewünschten Eigenschaften durch gezielte Vermehrung herausisoliert (separiert). Die verschiedenen Moleküle konkurrieren miteinander und die Moleküle, welche die gestellte Anforderung beziehungsweise Aufgabe am besten erfüllen, werden selektiert und weiter vermehrt. Anschließend werden die ausgewählten und vermehrten Moleküle erneut einer Selektion, unter variierten und meist verschärften Bedingungen, unterzogen. Danach werden wiederum nur die besten Moleküle erneut gezielt vermehrt.
Durch den wiederholten Kreislauf von
- Selektion bindender RNA-Moleküle, (Selektion)
- Abtrennung nicht bindender Sequenzen, (Separation)
- enzymatischer Vervielfältigung der mit dem Zielmolekül interagierenden RNA-Liganden (Amplifikation) und
- (optionaler) Mutation,
erhält man üblicherweise Sequenzen, die sehr spezifisch und hochaffin an den gewünschten Targets binden.[5] Diese evolutionäre Vorgehensweise ist besonders leicht mit Molekülen wie DNA oder RNA durchzuführen, da sie mit Hilfe der Polymerase-Kettenreaktion relativ einfach vermehrbar sind. Der Kreislauf Selektion – Separation – Amplifikation (– Mutation) wird bis zu zwölfmal wiederholt.
Die DNA/RNA-Bibliothek
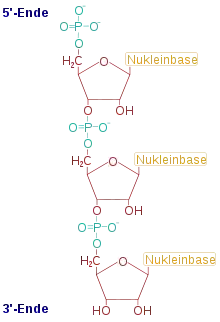
Die einzelsträngige DNA-Ausgangsbibliothek (ssDNA) wird meist durch chemische DNA-Synthesen mittels automatisierter Festphasensynthese hergestellt. Dabei wird ein DNA-Synthesizer so eingestellt, dass nicht ein Nukleotid an einer bestimmten Stelle gekuppelt wird, sondern eine Mischung aus allen vier Nukleobasen. Die randomisierte Sequenz wird in der Mitte zwischen bekannten Sequenzen hergestellt, die für die Primererkennung notwendig sind.[6]
Der durch PCR erzeugte doppelsträngige DNA- und in RNA umgeschriebene Pool kann nun im nächsten Schritt des Selektionsprozesses mit dem gewünschten Zielmolekül, unter sorgfältig ausgewählten Bedingungen, inkubiert werden.
Die Ausgangsbibliothek enthält für gewöhnlich einen Bereich von 20 bis 220 randomisierten Nukleotiden.[7] An den Enden befinden sich konstante primerbindende 3'- bzw. 5'-Sequenzen. Im konstanten Bereich liegt meist eine Promotorsequenz, die für die In-vitro-Transkription der RNA unerlässlich ist.[8]
Zwischen den beiden konstanten Enden befindet sich die eigentliche Zufallssequenz aus n-Nukleotiden, was 4n verschiedene mögliche Sequenzen ergibt. Rein rechnerisch sind aus einer Zufallssequenz mit n Nukleotid-Elementen:
- n = 25 eine Bibliothek aus ca. 1015 Sequenzen,
- n = 50 eine Bibliothek aus ca. 1030 Sequenzen,
- n = 75 eine Bibliothek aus ca. 1045 Sequenzen,
- n = 100 eine Bibliothek aus ca. 1060 Sequenzen möglich.[4]
Ein Mol, das heißt 6,022·1023 Moleküle eines 23mers – eine Nukleotidsequenz mit 23 randomisierten Nukleobasen – wiegt ca. 7,5 kg. Um statistisch ein Molekül von jeder möglichen Variante zu haben, benötigt man 75 µg Oligonukleotid. Für eine 50-fache statistische Deckung benötigt man lediglich 3,75 mg Oligonukleotid.
Völlig anders dagegen ist die Situation bei den längerkettigen Sequenzen. Für eine einfache statistische Deckung würde man, nach Abzug der Primer, beim 40mer 13 kg, beim 50mer 16 kg und beim 100mer 32 kg benötigen.[4] Dies ist ab der 40mer praktisch nicht mehr möglich.
Der Nachteil langer Zufallssequenzen ist, dass je länger die Zufallssequenz wird, desto kleiner wird der Ausschnitt der möglichen Sequenzen, den eine Probe im mg-Maßstab repräsentiert. Der Vorteil langer Zufallssequenzen ist dagegen, dass ein einziges 100mer bereits 75 verschiedene 25mer Sequenzen bzw. 50 verschiedene 50mer Sequenzen enthält. Die kann das Selektionsverfahren deutlich beschleunigen.[4]
Selektion
Die Inkubation zwischen den Nukleotid-Sequenzen und den Zielstrukturen (Targets) kann im immobilisierten Zustand oder in Lösung stattfinden. Die Wechselwirkungskräfte zwischen der Vielzahl an Nukleotid-Sequenzen und den Zielstrukturen sind dabei sehr unterschiedlich. Die auf dem Schlüssel-Schloss-Prinzip basierende Bindung der Nukleotide ist von ihrer Sekundärstruktur extrem stark abhängig. Die Sekundärstruktur hängt wiederum sehr stark von der Sequenz, das heißt der Reihenfolge der Nukleobasen Adenin, Guanin, Cytosin, Thymin (im Falle von DNA) bzw. Uracil (im Fall von RNA) ab.
Als Sekundärstrukturen werden beispielsweise Helices, Triple-Helices, Haarnadel-Schleifen, Pseudoknoten und G-Quartette beschrieben.[9][10][11][12]
Separation
Durch unterschiedliche Waschschritte werden die an den Targets nur schwach oder gar nicht gebundenen Sequenzen von den gebundenen Sequenzen abgetrennt (separiert). Üblicherweise erfolgt die Abtrennung mit Hilfe der Affinitätschromatografie. Die gebundenen Sequenzen werden eluiert und mit der PCR vermehrt.[13]
Amplifikation
Die Amplifikation erfolgt durch eine reverse Transkription (nur bei RNA) und die Polymerase-Kettenreaktion (PCR). Daraus entsteht ein Pool von DNA bzw. RNA-Molekülen mit – im Vergleich zum ursprünglichen Molekülpool – verbesserten Bindungseigenschaften zum Liganden. Mit diesem Schritt schließt sich der Kreislauf und wird nun für gewöhnlich acht bis zwölfmal wiederholt, bis ein gewünschtes Aptamer erhalten wird, oder nur wenig verschiedene Sequenzen angereichert sind. Durch Klonierung und Sequenzierung resultieren nach Abschluss der systematischen Evolution monoklonale Aptamere. Als mögliche Zielmoleküle (Targets) kommen verschiedenste Moleküle wie einfache organische Verbindungen, Proteine oder ganze Zellen in Frage.[5]
Mutation
Wie bereits bei der Molekülbibliothek beschrieben, ist eine komplette Randomisierung eines 100meren Oligonukleotid mit 1060 (=4100) Varianten nicht realisierbar. Man kann nur ca. 1016 Moleküle produzieren. Die aus diesem limitierten Pool isolierten aktivsten Oligonukleotide, können zu einer weiteren Verbesserung durch mutagene PCR leicht verändert werden. Man spricht in diesem Fall auch von Nachrandomisieren. Dabei werden die PCR-Bedingungen so eingestellt, dass die Polymerase während des Kopiervorganges Fehler macht. Eine Fehlerrate von beispielsweise 1 pro 20 Basenpaaren kann durch die Wahl der Bedingungen eingestellt werden.[6]
Das Nachrandomisieren empfiehlt sich erst nach mehreren Durchläufen des Selektions-Separations-Amplifikations-Zyklus. Es ist genau genommen kein gesonderter Prozessschritt, sondern eine Modifizierung des Amplifikations-Schrittes.
Geschichte
Die SELEX-Methode wurde 1990 zeitgleich von Larry Gold[2] und Andrew E. Ellington[1] entwickelt. Beide Ansätze basieren auf dem Prinzip der selektiven Erkennung von Zielstrukturen, vermittelt durch die 3-dimensionale Form von einzelsträngigen Nukleinsäuren. Gold verwendete dabei einsträngige DNA (single strand DNA, ssDNA), Ellington dagegen RNA mit definierter Oberflächenstruktur.[14]
Bedeutung des Verfahrens
Die Selektion neuer Enzyme auf RNA- und DNA-Basis, den sogenannten Ribozymen, ist in den Fokus der Forschung gerückt. Die katalytisch aktive RNA, die sich in spezifische dreidimensionale Formen faltet, kann wie ein auf Proteinen basierendes Enzym Reaktionen katalysieren. Zudem können die Ribozyme wie ein Antikörper hochspezifisch kleine Moleküle oder Proteine molekular binden.
Die Herstellung dieser Aptamere genannten, hochspezifischen Rezeptoren auf DNA- oder RNA-Basis, die an medizinisch relevante Targets binden können, ist das Ziel von SELEX.[6]
Das Verfahren wird genutzt, um Aptamere mit extrem hoher Bindungsaffinität an unterschiedliche Targets zu entwickeln. Auch kleine Moleküle wie ATP,[15] Adenosin[16][17] und Proteine, wie beispielsweise Prionen[18] und Signalmoleküle wie Vascular Endothelial Growth Factor (VEGF),[19] sind Targets für Aptamere.
Im Bereich der Onkologie sind Aptamere sehr vielversprechende Liganden.[20]
Das an VEGF bindende Aptamer Pegaptanib ist für die Behandlung der altersabhängigen Makula-Degeneration (AMD)[19][21] zugelassen.
Es ist allerdings zu beachten, dass extrem hohe Bindungsaffinitäten im sub-nanomolaren Bereich nicht unbedingt die Spezifität zum Zielmolekül erhöhen.[22] Die sogenannte Off-Target-Bindung hat signifikante klinische Auswirkungen.
Die Entwicklungs- und Herstellungskosten für Biosensoren auf Aptamerbasis werden in der Literatur als erheblich niedriger eingeschätzt als auf Basis von Proteinen (z. B. Antikörper).[4]
Molekülmodifikationen
Die Verwendung von 2’-Fluor- oder 2’-Amino-Mononukleotid-Triphosphat modifizierten RNA-Bibliotheken ermöglicht die Gewinnung von Ribonuklease-resistenten Aptameren.[23] Derart modifizierte Aptamere haben das Potenzial, auch in biologischen Flüssigkeiten (Blut, Urin) als Sonde zu fungieren.[14][24]
Einzelnachweise
- A. D. Ellington u. a.: In vitro selection of RNA molecules that bind specific ligands. in Nature, 346/1990, S. 818–822
- C. Tuerk u. a.: Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. In: Science 249/1990, S. 505–510.
- A. D. Ellington, J. W. Szostak: Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. In: Nature 355/1992, S. 850–852
- Landesumweltamt NRW: 2006: Antibiotika, Resistenzen und Bakterien in Kläranlagen. (Seite nicht mehr abrufbar, Suche in Webarchiven) Info: Der Link wurde automatisch als defekt markiert. Bitte prüfe den Link gemäß Anleitung und entferne dann diesen Hinweis. (PDF; 4,2 MB) 2006
- Sabine Kainz: Selektion und Charakterisierung von Aptameren, spezifisch für das Virus der Infektiösen Bursitis. Dissertation, Hamburg 2005
- Universität Marburg: Anwendung der PCR in der in vitro random selection (Memento des Originals vom 3. September 2016 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- D. P. Bartel, J. W. Szostak: Isolation of new ribozymes from a large pool of random sequences. In: Science 261/1993, S. 1411–1418
- A. A. Beaudry, G. F. Joyce: Directed evolution of an RNA enzyme. In: Science 257/1992, S. 635–641
- N. Leontis u. a.: The non-Watson-Crick base pairs and their associated isostericity matrices. In: Nucleic Acids Research, 30/2002, S. 3497–3531.
- B. E. Eaton u. a.: Ribonucleosides and RNA. In: Annual Review of Biochemistry. 64/1995, S. 837–863.
- F. Jiang u. a.: Structural basis of RNA folding and recognition in an AMP-RNA aptamer complex. In: Nature 382/1996, S. 183–186.
- Woese CR, Architecture of ribosomal RNA: constraints on the sequence of "tetra-loops. In: Proceedings of the National Academy of Sciences. 87/1990, S. 8467–8471.
- F. Jarosch u. a.: In vitro selection using a dual RNA library that allows primerless selection. In: Nucleic Acids Research. 34/2006, S. 86.
- Michael Blank: Systematische Evolution von DNA-Aptameren zur Charakterisierung und Diagnostik von pathologisch veränderten Endothelzellen in Tumoren und entzündlichen Regionen des Rattenhirns. Tübingen 2002, DNB 965284956, urn:nbn:de:bsz:21-opus-5841 (Dissertation, Universität Tübingen).
- T. Dieckmann u. a.: Solution structure of an ATP-binding RNA aptamer reveals a novel fold. In: RNA 2/1996, S. 628–640.
- D. E. Huizenga, J. W. Szostak: A DNA aptamer that binds adenosine and ATP. In: Biochemistry. 34/1995, S. 656–665.
- D. H. Burke, L. Gold: RNA aptamers to the adenosine moiety of S-adenosyl methionine: structural inferences from variations on a theme and the reproducibility of SELEX. In: Nucleic Acids Research. 25/1997, S. 2020–2024.
- R. Mercey u. a.: Fast, reversible interaction of prion protein with RNA aptamers containing specific sequence patterns. In: Archives of Virology. 151/2006, S. 2197–2214.
- H. Ulrich u. a.: DNA and RNA aptamers: from tools for basic research towards therapeutic applications. In: Combinatorial Chemistry & High Throughput Screening. 9/2006, S. 619–632.
- C. S. Ferreira u. a.: DNA aptamers that bind to MUC1 tumour marker: design and characterization of MUC1-binding single-stranded DNA aptamers. In: Tumor Biology. 27/2006, S. 289–301.
- D. Vavvas, D. J. D'Amico: Pegaptanib (Macugen): treating neovascular age-related macular degeneration and current role in clinical practice. In: Ophthalmology Clinics of North America., 19/2006, S. 353–360.
- J. M. Carothers u. a.: Aptamers selected for higher-affinity binding are not more specific for the target ligand. In: Journal of the American Chemical Society. 128/2006, S. 7929–7937.
- B. Eaton u. a. in Annual Review of Biochemistry. 64/1994, S. 837–863
- W. Pieken in Science, 253/1991, S. 314–317
Literatur
- Michael G. Radermacher: Entwicklung eines DNS-Aptamers gegen das aktivierte Integrin GP IIB/IIIA. (PDF; 4,6 MB) Dissertation, 2007, Albert-Ludwigs-Universität Freiburg i. Br.
- Sylvia Ehses: Isothermale in vitro Selektion und Amplifikation zur Untersuchung von Evolutionsvorgängen. Dissertation, 2005, Ruhr-Universität Bochum
- Claudia Catharina Gauglitz: Direkte und nichtkompetitive Bestimmung von Bindungskonstanten mit fluoreszierenden RNA-Liganden. Dissertation, 2000, FU Berlin
- Mark Matzas: Selektion und Identifikation von Peptid Nukleinsäuren. Dissertation, 2005, Universität Hamburg