Hereditäres non-polypöses kolorektales Karzinom
Das hereditäre non-polypöse Kolonkarzinom (korrekte Bezeichnung eigentlich: hereditäres nicht-Polyposis-assoziiertes kolorektales Karzinom) (HNPCC) ist eine erbliche Darmkrebsform ohne Polyposis, d. h. ohne Auftreten von vielen Polypen im Darm. Uneinheitlich ist jedoch in der Nomenklatur, ob mit HNPCC ausschließlich Fälle mit einer nachgewiesenen Mutation in einem der bekannten HNPCC-assoziierten Gene bezeichnet werden (klassisches Lynch-Syndrom), oder auch erbliche Darmkrebsformen mit gleicher oder ähnlicher Erscheinung, aber bislang unklarer Ursache. Letztere werden deswegen mitunter unter dem Terminus Familiäres Kolorektalkarzinom Typ X (FCCTX) zusammengefasst.[1] Nicht zu verwechseln mit dem HNPCC ist die Familiäre adenomatöse Polyposis (FAP), eine erbliche Darmkrebsform mit einem massenhaften Auftreten von Polypen.
| Klassifikation nach ICD-10 | |
|---|---|
| C18.9 | Bösartige Neubildung: Kolon, nicht näher bezeichnet |
| Z80.0 | Bösartige Neubildung der Verdauungsorgane in der Familienanamnese |
| ICD-10 online (WHO-Version 2019) | |
Epidemiologie
Das hereditäre Dickdarm-Karzinom ohne Polyposis (HNPCC) ist die häufigste erbliche Darmkrebsform und betrifft etwa drei Prozent der Darmkrebsfälle.[2][3] Seine Prävalenz in der Allgemeinbevölkerung liegt bei etwa 1 zu 300[4] bis 500[5], womit es die häufigste Krebsdisposition überhaupt darstellt.
Ursache
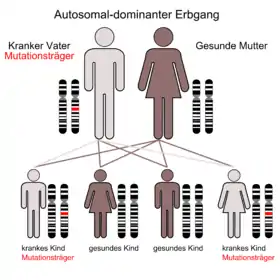
Mittlerweile sind 5 Gene (MSH2, MSH6, MLH1, PMS2, und EPCAM) bekannt, die, insofern eine pathogene Mutation vorliegt, zum Auftreten eines hereditären non-polypösen Karzinoms führen können. Diese Gene kodieren für Proteine aus der Gruppe der sogenannten DNA-Mismatch-Reparaturproteine, deren Aufgabe es ist, eventuelle Fehler bei der Replikation der DNA im Rahmen der Zellteilung zu erkennen und zu beseitigen. Ausnahme davon ist EPCAM, das durch seine Lage direkt vor MSH2 dessen Expression beeinflussen kann. Die Vererbung ist autosomal dominant, das bedeutet, dass Betroffene die Erkrankung mit einer Wahrscheinlichkeit von 50 % unabhängig vom Geschlecht an ihre Kinder weitergeben und dass die Erkrankung mit sehr großer Wahrscheinlichkeit auch mit nur einem betroffenen Allel manifest wird.
Darüber hinaus wurden inzwischen zwei weitere Gene identifiziert, TGFBR2[6] und MLH3[7], deren Funktionsverlust die HNPCC-Typen 6 und 7 verursacht. Im Unterschied zu den klassischen HNPCC-Typen scheinen Tumoren dieser Ätiologie nicht mit einer Mikrosatelliteninstabilität verknüpft zu sein, was auf einen anderen Pathomechanismus hindeutet. Darüber hinaus ist hier wegen einer bislang zu geringen Fallzahl nicht geklärt, wie penetrant pathogene Varianten dieser Gene sind, anscheinend tritt die Erkrankung mit einer geringeren Wahrscheinlichkeit und in einem eher höheren Alter auf. Jedoch variiert die Penetranz auch innerhalb der klassischen HNPCC-Gene: Mutationen in den Genen MLH1 und MSH2 beeinträchtigen die DNA-Reparatur stärker als Mutationen in MSH6 und PMS2[8] und gelten daher als Hochrisikogene.
Tumorspektrum
Im Allgemeinen haben Menschen bei denen ein HNPCC diagnostiziert wurde ein erhöhtes Lebenszeitrisiko für verschiedene Krebserkrankungen u. a. des Dickdarms, des Magens, des Urogenitaltraktes, des Dünndarms, der Gallenwege, des ZNS und der Haut. Bei weiblichen Individuen besteht zusätzlich ein deutlich erhöhtes Lebenszeitrisiko für Gebärmutterschleimhaut- und Eierstockkrebs. Das genaue Lebenszeitrisiko ist allerdings abhängig vom zugrunde liegenden Gendefekt.
Eine HNPCC-Variante stellt das Muir-Torre-Syndrom (MRTES) dar, bei dem zusätzlich Talgdrüsenadenome und Keratoakanthome auftreten.
Molekularbiologie des hereditären non-polypösen Karzinoms
Charakteristischerweise werden mittels molekularbiologischer Methoden bei Betroffenen Veränderungen der Mikrosatellitenmarker nachgewiesen. Bei Patienten mit einem hereditären non-polypösen Karzinom findet man innerhalb eines betroffenen Individuums eine Sequenzlängendifferenz zwischen Tumor und gesundem Gewebe als Hinweis auf eine fehlerhafte DNA-Replikation. Man bezeichnet dieses Phänomen als Mikrosatelliteninstabilität. Diese tritt jedoch nicht bei allen Formen auf (siehe oben).
Diagnosekriterien
Amsterdam II-Kriterien
Alle Kriterien müssen erfüllt sein.
- mindestens drei Familienangehörige mit HNPCC-assoziiertem Karzinom (Kolon/Rektum, Endometrium, Dünndarm, Nierenbecken/Ureter)
- einer davon Verwandter ersten Grades der beiden anderen
- Erkrankungen in mindestens zwei aufeinanderfolgenden Generationen
- mindestens ein Patient mit der Diagnose eines Karzinoms vor dem 50. Lebensjahr
- Ausschluss einer FAP (Familiäre adenomatöse Polyposis)
Revidierte Bethesda-Kriterien
Mindestens ein Kriterium muss erfüllt sein.
Tumoren von Patienten sollten auf das Vorliegen einer Mikrosatelliten-Instabilität in folgenden Fällen untersucht werden:
- Patienten mit kolorektalem Karzinom vor dem 50. Lebensjahr.
- Patienten mit synchronen oder metachronen kolorektalen Karzinomen oder anderen HNPCC-assoziierten Tumoren (*), unabhängig vom Alter.
- Patienten mit kolorektalem Karzinom mit MSI-H Histologie (**) vor dem 60. Lebensjahr.
- Patient mit kolorektalem Karzinom (unabhängig vom Alter), der einen Verwandten 1. Grades mit einem kolorektalen Karzinom oder einem HNPCC-assoziierten Tumor vor dem 50. Lebensjahr hat.
- Patient mit kolorektalem Karzinom (unabhängig vom Alter), der mindestens zwei Verwandte 1. oder 2. Grades hat, bei denen ein kolorektales Karzinom oder ein HNPCC-assoziierter Tumor (unabhängig vom Alter) diagnostiziert wurde.
(*) zu den HNPCC-assoziierten Tumoren gehören Tumoren in: Kolorektum, Endometrium, Magen, Ovarien, Pankreas, Ureter oder Nierenbecken, Gallengang, Dünndarm und Gehirn (meist Glioblastome wie bei Turcot-Syndrom) sowie Talgdrüsenadenome und Keratoakanthome (bei Muir-Torre-Syndrom)
(**) Vorliegen von Tumor-infiltrierenden Lymphozyten, Crohn-ähnlicher lymphozytärer Reaktion, muzinöser/Siegelring-Differenzierung, oder medullärem Wachstumsmuster
Früherkennungsprogramm zur Vor- und Nachsorge
Vom Verbundprojekt „Familiärer Dickdarmkrebs“ wird ein lebenslanges Früherkennungsprogramm für HNPCC-Patienten empfohlen:[9]
Einmal jährlich zusätzlich zu den sonstigen Nachsorgen wie Blutuntersuchungen, okkultes Blut im Stuhl, Brust:
- Körperliche Untersuchung
- Abdomensonographie
- Komplette Koloskopie
- Gynäkologische Untersuchung auf Endometrium- und Ovarialkarzinom (inkl. transvaginaler Sonographie und Pipelle-Endometriumbiopsie)[10][11]
- Gastroduodenoskopie (ab dem 35. Lebensjahr)
Für Familienmitglieder ersten Grades gelten die gleichen jährlichen Untersuchungsempfehlungen zur Vorsorge, es sei denn, auf molekulargenetischer Ebene konnte der Nachweis erbracht werden, dass der Gendefekt nicht geerbt wurde. Der Beginn der Untersuchungen wird ab dem 25. Lebensjahr empfohlen, spätestens aber 5 Jahre vor dem zuerst in der Familie aufgetretenen Fall (Bsp.: jüngste Betroffene in der Familie war bei Auftreten der Erkrankung 25 Jahre alt, dann sollten Vorsorgeuntersuchungen für alle Familienmitglieder spätestens mit dem 20. Lebensjahr beginnen).
Einzelnachweise
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4119982/
- Lynch Syndrome–Associated Colorectal Cancer. In: NEJM.org. Abgerufen am 30. Dezember 2018 (englisch).
- Robert J. MacInnis, John L. Hopper, Polly A. Newcomb, Noralane M. Lindor, Yingye Zheng: Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. In: Cancer Epidemiology and Prevention Biomarkers. Band 26, Nr. 3, 1. März 2017, ISSN 1538-7755, S. 404–412, doi:10.1158/1055-9965.EPI-16-0693, PMID 27799157, PMC 5336409 (freier Volltext) – (aacrjournals.org [abgerufen am 30. Dezember 2018]).
- https://www.mgz-muenchen.de/files/mgz/Download/Diagnostische%20Schwerpunkte/Gastrointestinale%20Tumorerkrankungen%20(Uebersichtskarte).pdf
- Verena Steinke, Christoph Engel, Reinhard Büttner, Hans Konrad Schackert, Wolff H Schmiegel: Hereditary Nonpolyposis Colorectal Cancer (HNPCC)/Lynch Syndrome. In: Deutsches Ärzteblatt International. Band 110, Nr. 3, Januar 2013, ISSN 1866-0452, S. 32–38, doi:10.3238/arztebl.2013.0032, PMID 23413378, PMC 3566622 (freier Volltext).
- https://www.omim.org/entry/614331
- https://www.omim.org/entry/614385
- https://www.aerzteblatt.de/archiv/134013/Erblicher-Darmkrebs-ohne-Polyposis
- Verbundprojekt „Familiärer Dickdarmkrebs“ der Deutschen Krebshilfe
- R. Schneider u. a.: Das Lynch-Syndrom – Epidemiologie, Klinik, Genetik, Screening, Therapie. In: Zeitschrift für Gastroenterologie. 2012; 50, S. 217–225.
- W. Schmiegel u. a.: S3-Leitlinie "Kolorektales Karzinom". In: Zeitschrift für Gastroenterologie. 2008, 46, S. 1–73 (Archivlink (Memento des Originals vom 29. Dezember 2009 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.)