Aluminiumorganische Verbindungen
Aluminiumorganische Verbindungen, auch Aluminiumorganyle genannt, sind chemische Verbindungen, die eine Bindung zwischen Kohlenstoff und Aluminium besitzen. Sie zählen zu den wichtigsten metallorganischen Verbindungen.[1][2] Beispielhafte aluminiumorganische Verbindungen sind das Dimer Trimethylaluminium, das Monomer Triisobutylaluminium und die Titan-Aluminium-Verbindung Tebbe-Reagenz. Das Verhalten von aluminiumorganischen Verbindungen wird beeinflusst durch die Polarität der C-Al-Bindung und die hohen Lewis-Acidität des meist dreifach koordinierten Aluminiumatoms. Technisch werden diese Verbindungen vor allem für die Herstellung von Polyolefinen verwendet.
Geschichte
Die erste aluminiumorganischen Verbindung (C2H5)3Al2I3 wurde im Jahre 1859 entdeckt.[3] Jedoch waren Aluminiumorganyle von geringer Bedeutung, bis Karl Ziegler und Kollegen 1950 die Direktsynthese von Trialkylaluminiumverbindungen entdeckten und diese zur katalytische Olefin-Polymerisation verwenden konnten. Diese Entdeckung führte letztlich zur Vergabe des Nobelpreises an Ziegler.
Struktur und Bindungsverhältnisse
Aluminium(III)-Verbindungen
Aluminiumorganische Verbindungen besitzen meist drei- oder vierfach koordinierte Aluminium-Zentren, obwohl auch höhere Koordinationszahlen mit anorganischen Liganden (wie etwa Fluorid) beobachtet werden. In Übereinstimmung mit generellen Trends sind vierfach koordinierte Aluminium-Verbindungen bevorzugt tetraedrisch. Im Vergleich zum Boratom ist das Aluminium-Atom größer, sodass vier Kohlenstoffliganden einfacher Platz finden. Aus diesem Grund sind Triorganoaluminium-Verbindungen in der Regel Dimere, mit einem Paar von Alkyl-Brückenliganden, beispielsweise Al2(C2H5)4(μ-C2H5)2. Trotz des gängigen Namens Triethylaluminium enthält diese Verbindung von zwei Aluminiumzentren und sechs Ethylgruppen. Wenn in der aluminiumorganischen Verbindung Hydride oder Halogenide enthalten sind, neigen diese kleineren Liganden dazu, die Brückenplätze zu besetzen. Dreifache Koordinations tritt auf, wenn die R-Gruppen sperrig sind, z. B. Al(Mes)3 (Mes = 2,4,6-Me3C6H2 or Mesitylen) oder Isobutyl.[4]
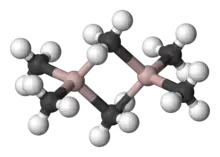
Ligandenaustausch in Trialkylaluminiumverbindungen
Trialkylaluminium-Dimere unterliegen meist einem dynamischen Gleichgewichte; durch dieses tauschen sie sowohl verbrückende und terminale Liganden, wie auch die Liganden untereinander. Der Methyl-Austausch ist auch in nicht-koordinierenden Lösungsmitteln recht rasch, wie durch Protonen-NMR-Spektroskopie bestätigt wurde. So besteht das 1H-NMR-Spektrum von Me6Al2 bei −25 °C aus 2 Signalen im 1:2-Verhältnis, wie aufgrund der Struktur zu erwarten ist. Bei 20 °C ist hingegen nur noch ein Signal sichtbar, da der Austausch von verbrückenden und terminalen Methylgruppen zu schnell ist, um durch NMR-Spektroskopie noch aufgelöst zu werden. Die hohe Lewis-Acidität der monomeren Spezies steht im Verhältnis zur Größe des Al(III)-Zentrums und seiner Tendenz, die Oktett-Konfiguration zu erreichen.
Niedervalente aluminiumorganische Verbindungen
Die erste aluminiumorganischen Verbindung mit einer Al-Al-Bindung, (((Me3Si)2CH)2Al)2 (ein Dialan), wurde 1988 gefunden. Verbindungen dieser Art werden typischerweise durch Reduktion von Dialkylaluminiumchloriden mit metallische Kalium gewonnen:[5]
- (R2AlCl)2 + 2 K → R2Al–AlR2 + 2 KCl
Eine weitere bemerkenswerte Gruppe von Alanen sind Tetraalane, bestehend aus vier Al(I)-Zentren. Diese Verbindungen besitzen einen Tetrahedran-Kern, gezeigt durch (Cp*Al)4 und ((Me3Si3C)Al)4. Der Cluster [Al12(i-Bu)12]2− wurde aus ähnlichen Untersuchungen über die Reduktion von aluminiumorganische Verbindungen gefunden. Dieses Dianion nimmt eine ikosaederförmige Struktur ein, die an Dodecaborat ([B12H12]2−) erinnert. Sein formaler Oxidationszustand beträgt weniger als eins.
Darstellung
Aluminiumorganische Verbindungen werden analog zu den siliciumorganischen Verbindungen in erster Linie durch folgende drei Verfahren gebildet:
durch oxidative Addition von Organylhalogeniden an Aluminium (im sogenannten Direktverfahren)
- 2 Al + 3 RX → RAlX2 / R2AlX
durch nukleophile Substitution von Aluminiumhalogeniden durch Organylanionen (Metathese)
- R2Al-X + R− → R2Al-R + X−
oder durch Insertion von Alkenen oder Alkinen in die Al-H-Bindung von Organylalanen (Hydroaluminierung):
- R2Al-H + H2C=CH2 ⇌ Al–CH2-CH2-H.
Die Ummetallierung von Quecksilberorganylen wird nur selten verwendet:
- 2 Al + 3 HgR2 → 2 AlR3 + 3 Hg.
Aus Alkylhalogeniden und Aluminium (Direktverfahren)
Einfache Trialuminiumalkyle Al2R6 werden technisch im Hüls-Verfahren hergestellt. Das Verfahren besitzt nur für Verbindungen mit R = Me, Et (Trimethylaluminium und Triethylaluminium) eine Bedeutung. Das zweistufige Verfahren beginnt mit der Alkylierung von Aluminiumpulver:
- 2 Al + 3 EtCl → Et3Al2Cl3 ⇌ Et4Al2Cl2 + Et2Al2Cl4
- Aluminium und Chlorethan reagieren zu Ethylaluminiumchlorid, welches im Gleichgewicht mit den Disproportionierungsprodukten steht.
Die Reaktion erinnert an die Synthese von Grignard-Verbindungen. Das Produkt, Et3Al2Cl3, wird Ethylaluminiumsesquichlorid genannt. „Sesquichlorid“ bezieht sich dabei auf das durchschnittliche Cl:Al-Verhältnis von 1,5. Das Sesquichlorid disproportioniert in einer Gleichgewichtsreaktion; das Et2Al2Cl4 ist im Reaktionsgemisch unerwünscht und wird durch Zugabe von Natriumchlorid komplexiert und ausgefällt:
- Et2Al2Cl4 + 2 NaCl → 2 Na[EtAlCl3]s ↓.
Das Et4Al2Cl2 wird durch Destillation abgetrennt mit Natrium in das Triorganylaluminium-Derivate reduziert:
- 3 Et4Al2Cl2 + 6 Na → 2 Et6Al2 + 6 NaCl + 2 Al.
Als Gesamtgleichung ergibt sich:
- Al + 3 RCl + 3 Na → AlR3 + 3 NaCl.[6]
Hydroaluminierung
Bei der Hydroaluminierung (auch Ziegler-Prozess genannt) reagieren in Summe Alkene (hier: Ethen) mit Aluminium und Wasserstoff. Dazu werden zwei einzelne Reaktionen kombiniert. Zunächst reagieren Aluminium, Wasserstoff und Aluminiumtriorganyle zum Dialkylaluminiumhydrid (die "Vermehrung"):
- Al + 3/2 H2 + 2 AlEt3 → 3 Et2AlH
Im dann folgenden Schritt werden diese Hydride mit dem Alken in einer Hydroaluminierung umgesetzt (in der "Anlagerung"):
- 3 Et2AlH + 3 CH2=CH2 → 3 AlEt3
Als Gesamtgleichung ergibt sich:
- Al + 3/2 H2 + Alken → AlR3
Da die Anlagerung reversibel ist, lassen sich auf diesem Weg mit einer nachfolgenden Carbaluminierung zahlreiche weitere Trialkylorganyle technisch herstellen, z. B. Tris(n-octyl)aluminium aus Tris(/-butyl)aluminium (siehe Kapitel Dehydro-, Hydro- und Carbaluminierung).
Laborpräparation
Da viele Aluminiumorganyle durch industrielle Herstellung zu geringen Kosten kommerziell verfügbar sind, beschränkt sich die Herstellung im Labor auf spezielle Verbindungen. Diese werden meist durch Metathese oder Transmetallierung hergestellt.
Metathese von Aluminiumtrichlorid mit RLi oder RMgX gibt die Trialkyl-:
- AlCl3 + 3 BuLi → Bu3Al + 3 LiCl
Die Transmetallierung läuft wie folgt ab:
- 2 Al + 3 HgPh2 → 2 AlPh3 + 3 Hg
Reaktionen
Die hohe Reaktivität der aluminiumorganische Verbindungen gegenüber Elektrophilen wird der Ladungstrennung zwischen dem Aluminium- und dem Kohlenstoffatom zugeschrieben.
Lewis-Acidität
Aluminiumorganische Verbindungen sind harte Säuren und reagieren leicht mit harten Basen wie Pyridin, THF und tertiären Aminen. Die Produkte sind tetraedrisch am Al-Zentrum.
Dehydro-, Hydro- und Carbaluminierung
Triorganylalane AlR3 zerfallen leicht in einer Dehydroaluminierung:
- R2Al–CR2-CR2H ⇌ R2Al–H + R2C=CR2
Es handelt sich um eine Gleichgewichtsreaktion, Alkene lassen sich in einer Hydroaluminierung an Di- oder Monoorganylalane (RAlH2 oder R2AlH) anlagern. Die Anlagerung verläuft stereoselektiv (cis) und recht regioselektiv (anti-Markownikow), aber wenig chemoselektiv (es werden neben C-C-Mehrfachbindungen auch solche zu anderen Elementen angegriffen; C=Y und C≡Y). Die Bereitschaft zur Hydroaluminierung wächst nach:
- RCH=CHR < R2C=CH2 < RCH=CH2 < CH2=CH2
Zudem reagieren Triorganylalane in der Carbaluminierung. Hierbei schiebt sich ein Alken oder Alkin in die Al-C-Bindung ein:
- R2Al–Et + n H2C=CH2 → R2Al–(CH2–CH2)n–Et.
Durch sukzessiv aufeinander folgende Insertionen ist die Bildung langer Kohlenstoffketten am Aluminium möglich (bis zu 200 Kohlenstoff-Atome). Als Begrenzung wirkt die Konkurrenz zwischen Dehydroaluminierung und Carbaluminierung; im Gegensatz zur Hydroaluminierung ist die Carbaluminierung jedoch nicht reversibel. Das Geschwindigkeitsgesetz der Reaktion folgt dabei:
- v = k[(Et3Al)2]l/2*[Alken].
Es wird angenommen, dass der Mechanismus zur Carbaluminierung über die Bildung eines Aluminiumorganyl-Alken-Komplexes verläuft, der sich in einem zweiten Schritt zum Produkt umlagert:
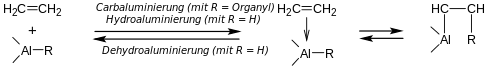
Auch die Hydroaluminierung (Einschub eines Alkens in eine Aluminium-Wasserstoff-Bindung) verläuft nach diesem Mechanismus. Jedoch ist der Mechanismus nur für den Fall der Hydroaluminierung reversibel.
Technisch ist die Carbaluminierung zunächst für die Herstellung von Vorstufen biologisch abbaubarer Tenside H3OS–O–(CH2–CH2)n–H (mit n etwa 13) von Bedeutung. Bei der Reaktion des Aluminiumorganyls mit Sauerstoff entstehen zunächst die Alkoholate R2Al–O–(CH2–CH2)n–H und nach Reaktion mit Wasser langkettige, unverzweigte Alkohole HO(CH2–CH2)n–H.
Elektrophile
Die Al-C-Bindung ist so stark polarisiert, dass der Kohlenstoff stark basisch ist. Säuren setzen Alkane in der Reaktion frei. Zum Beispiel geben Alkoholen Alkoholate:

Eine Vielzahl von Säuren kann auf diese Weise gebildet werden. Mit Aminen werden Amidoderivate gebildet. Mit Kohlendioxid bilden Trialkylaluminiumverbindungen Dialkylaluminium-Carboxylate:

Die Umwandlung erinnert an die Carbonisierung von Grignard-Reagenzien.
Mit Sauerstoff erhält man die entsprechenden Alkoxide, die zu Alkoholen hydrolysiert werden können:

Ein strukturell charakterisiertes Organoaluminium-Peroxid ist .[7]

Alkenpolymerisation
Technisch werden aluminiumorganische Verbindungen als Katalysatoren für die Alken-Polymerisation von Polyolefinen eingesetzt, beispielsweise der Katalysator Methylaluminoxan.
Eigenschaften
Die Aluminiumalkyle sind farblose Flüssigkeiten. Aluminiumalkyle mit kurzen Alkylketten R entflammen an Luft spontan und reagieren in Kontakt mit Wasser explosiv; solche mit langen Resten sind weniger reaktiv. Derivate mit usw. verhalten sich ebenfalls deutlich weniger reaktiv. Aluminiumorganyle reagieren mit den meisten Lösungsmitteln (Ausnahme: gesättigte und aromatische Kohlenwasserstoffe), der Kontakt mit Chloroform () kann sogar zu Explosionen führen. Die thermische Spaltung durch Dehydroaluminierung beginnt für Aluminiunorganyle mit β-verzweigten Alkylresten bei 80 °C, für Aluminiumorganyle mit n-Alkylresten bei 120 °C.





Einzelnachweise
- D. F. Shriver, P. W. Atkins: Inorganic Chemistry. Oxford University Press, 2006, ISBN 978-0-19-926463-6.
- M. Witt, H. W. Roesky: Organoaluminum chemistry at the forefront of research and development. (Memento vom 6. Oktober 2014 im Internet Archive) In: Curr. Sci. 78, 2000, S. 410.
- W. Hallwachs, A. Schafarik: Ueber die Verbindungen der Erdmetalle mit organischen Radicalen In: Liebigs Ann. Chem. 1859, 109, S. 206–209, doi:10.1002/jlac.18591090214.
- Christoph Elschenbroich: Organometallics. VCH, Weinheim 2006, ISBN 978-3-527-29390-2.
- W. Uhl: Organoelement Compounds Possessing Al–Al, Ga–Ga, In–In, and Tl–Tl Single Bonds. In: Adv. Organomet. Chem.. 51, 2004, S. 53–108. doi:10.1016/S0065-3055(03)51002-4.
- Michael J. Krause, Frank Orlandi, Alfred T. Saurage, Joseph R. Zietz Aluminum Compounds, Organic. In: Ullmann's Encyclopedia of Industrial Chemistry. Wiley-VCH Weinheim 2005, doi:10.1002/14356007.a01_543
- W. Uhl, B. Jana: A persistent alkylaluminum peroxide: surprising stability of a molecule with strong reducing and oxidizing functions in close proximity. In: Chemistry. Band 14, Nummer 10, 2008, S. 3067–3071, doi:10.1002/chem.200701916, PMID 18283706.