Pyruvatdecarboxylase
Pyruvatdecarboxylase ist ein homotetrameres Enzym (EC 4.1.1.1), das die Decarboxylierung von Brenztraubensäure zu Acetaldehyd und Kohlendioxid im Cytoplasma katalysiert. Unter anaeroben Bedingungen ist dieses Enzym ein Teil der Gärung, die in Hefen stattfindet, insbesondere in der Gattung Saccharomyces, um Ethanol durch Gärung zu produzieren. Die Umwandlung von Pyruvat in Acetaldehyd und Kohlenstoffdioxid durch Pyruvatdecarboxylase steht am Anfang dieses Prozesses.[2] Pyruvatdecarboxylase ist abhängig von den Cofaktoren Thiaminpyrophosphat und Magnesium. Das Enzym sollte nicht mit der Pyruvat-Dehydrogenase verwechselt werden, einer Oxidoreduktase (EC 1.2.4.1), die die oxidative Decarboxylierung von Pyruvat zu Acetyl-CoA katalysiert.
| Pyruvatdecarboxylase | ||
|---|---|---|
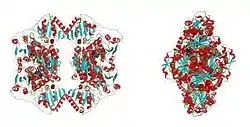
| ||
| Bändermodell (von oben, seitlich) des PDC-Tetramers der Bäckerhefe, mit Pyruvat/TPP/Mg++ als Kugeln, nach PDB 2VK1 | ||
| Sekundär- bis Quartärstruktur | Homotetramer | |
| Kofaktor | Thiaminpyrophosphat, Magnesium | |
| Enzymklassifikation | ||
| EC, Kategorie | 4.1.1.1, Lyase | |
| Reaktionsart | Decarboxylierung | |
| Substrat | Pyruvat | |
| Produkte | Acetaldehyd, Kohlendioxid | |
| Vorkommen | ||
| Übergeordnetes Taxon | Bakterien, Pilze, Pflanzen[1] | |

Hefe
In Hefen arbeitet Pyruvatdecarboxylase unabhängig während der anaeroben Vergärung und gibt als C2-Körper Acetaldehyd ab, sowie Kohlenstoffdioxid. Pyruvatdecarboxylase stellt einen Weg zur CO2-Eliminierung dar, das die Zelle abgibt. Das durch das Enzym erzeugte Ethanol dient als Antibiotikum, das eingesetzt wird, um konkurrierende Organismen zu beseitigen.[3] Das Enzym ist notwendig, um bei der Decarboxylierung von alpha-Ketosäuren zu helfen, weil im Übergangszustand am Carbonylkohlenstoffatom vermehrt negativen Ladungen angesammelt werden; das Enzym bietet damit die optimale Umgebung, damit sich Thiaminpyrophosphat und die alpha-Ketosäuren (Pyruvat) treffen können.[3]
Struktur
Pyruvatdecarboxylase ist als ein Dimer aus Dimeren aufgebaut, mit zwei aktiven Zentren, gebildet aus jeweils zwei Monomeren jedes Dimers. Das Enzym enthält eine beta-alpha-beta-Struktur, was zu parallelen beta-Faltblätter führt und 563 Rest-Untereinheiten in jedem Dimer. Die Monomere sind durch starke Wechselwirkungen zu Dimeren zusammengefügt, die Dimere interagieren aber nur lose miteinander, um ein lockeres Tetramer zu bilden.[3]
Das aktive Zentrum
Pyruvatdecarboxylase ist ein Homotetramer und besitzt somit vier aktive Zentren. Die aktiven Zentren befinden sich in einem Hohlraum im Kernbereich des Enzyms, wo Wasserstoffbrückenbindungen ausgebildet werden können und wo Pyruvat mit Thiaminpyrophosphat (TPP, s. Abbildung 2) reagiert. Jedes aktive Zentrum ist aus 20 Aminosäuren aufgebaut, einschließlich Glu-477 (trägt zur Stabilität des TPP-Rings bei) und Glu-51 (hilft bei der Cofaktor-Bindung). Diese Glutamate tragen auch dazu bei, das TPP-Ylid zu bilden, indem sie als Protondonator gegenüber dem substituierten TPP-Aminopyrimidinring wirken. Die Mikroumgebung um Glu 477 ist sehr unpolar, durch einen höheren pKa als gewöhnlich (normalerweise beträgt der pKA von Glu und Asp in kleinen Proteinen etwa 4,6).[4]
Die lipophilen Reste Ile-476, Ile-480 und Pro-26 tragen zur Unpolarität der Gegend um Glu-477 bei. Der einzige andere negativ geladene Rest, abgesehen vom TPP-Coenzym, ist Asp-28, das auch bei der Erhöhung des pKa von Glu-477 hilft. So muss die Umgebung des Enzyms die Protonierung der γ-Carboxygruppe von Glu-477 bei einem pH um 6 ermöglichen.[4]
Der Aminopyrimidinring am TPP deprotoniert, sobald er als Imin vorliegt, das C2-Atom von TPP, um so das nucleophile Ylid zu formen.[3] Dies muss in dieser Weise erfolgen, da das Enzym keine basischen Seitenketten besitzt, um das C2 von TPP zu deprotonieren. Eine Mutation im aktiven Zentrum in Zusammenhang mit Glu kann zur Ineffizienz oder Inaktivität des Enzyms führen. Diese Untätigkeit wurde in Experimenten gezeigt, in denen entweder die N1' und/oder 4'-Amino-Gruppe fehlten. In NMR-Analysen wurde festgestellt, dass, wenn TPP an das Enzym zusammen mit dem zum Substrat analogen Pyruvamide gebunden ist, die Rate der Ylid-Bildung größer als die des normalen Enzyms ist. Darüber hinaus verringert die Mutationsrate von Glu 51 zu Gln diese Rate deutlich.[3]
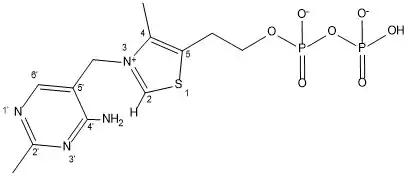
Ebenfalls enthalten sind Asp-444 und Asp-28, die das aktive Zentrum stabilisieren. Sie wirken als Stabilisatoren für das Mg2+-Ion, das sich in jedem aktiven Zentrum befindet. Um sicherzustellen, dass nur Pyruvat bindet, lösen zwei Cys-221 (mehr als 20 Angström von jedem Zentrum entfernt) und His-92 eine Konformationsänderung aus, die das Enzym, abhängig vom mit ihm interagierenden Substrat, hemmt oder aktiviert. Wenn das Substrat, das im aktiven Zentrum gebunden ist, Pyruvat ist, dann wird das Enzym durch eine Konformationsänderung dieser regulatorischen Site aktiviert.[5] Die Konformationsänderung beinhaltet eine 1,2 nucleophile Addition. Diese Reaktion, die Bildung eines Thioketals, wandelt das Enzym von der inaktiven in die aktive Form um.
Die Hemmung der Site wird ausgelöst durch einen Inhibitor bzw. ein Substrat-Analogon der Form XC6H4CH=CHCOCOOH, sowie durch Produkte der Decarboxylierung von Stoffen wie Zimtaldehyd. Weitere potenzielle Angriffsstellen für nucleophile Hemmer sind Cys-152, Asp-28, His-114, His-115 und Gln-477.[5]
Die normale katalytische Rate von Pyruvatdecarboxylase ist kcat = 10 s−1. Dagegen beträgt sie mit einer Glu-51-Mutation zu Gln 1,7 s−1.[3]
Mechanismus
Die Decarboxylierung von Pyruvat stellt die Herausforderung, dass eine negative Ladung an einem Carbonyl-Kohlenstoff, welche durch Eliminierung von CO2 entstünde, sehr instabil ist.[6]
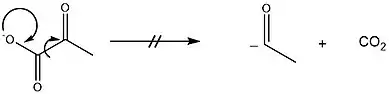
In der Pyruvatdecarboxylase-Reaktion fängt der Cofaktor TPP diese hohe Elektronendichte durch Delokalisierung auf (die gleiche "Strategie" findet man in der Pyruvatdehydrogenase-Reaktion[7]). Der entsprechende Carbonylkohlenstoff wird dafür zuvor zum Alkohol transformiert. Diese Reaktionsführung findet man auch in der Synthetischen Chemie, sogenannten Umpolungsreaktionen, wie zum Beispiel der Stetter-Reaktion oder der Benzoinaddition.
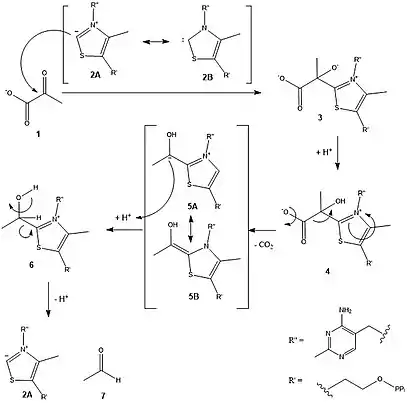
Das Proton am C2 des substituierten Thiazoliumrings (vgl. Abbildung 2) ist ausreichend azide, um durch den benachbarten Aminopyrimidinring deprotoniert zu werden. Der Reaktionsmechanismus der Deprotonierung ist in der untenstehenden Abbildung dargestellt. Dabei deprotoniert eine Base die Aminogruppe des Aminopyridinringes, was anschließend zur Bildung des Imins führt (Imin-Enamin-Tautomerie). Danach folgt ein interner Protonentransfer vom Thiazoliumring zur Aminogruppe.[8][9]
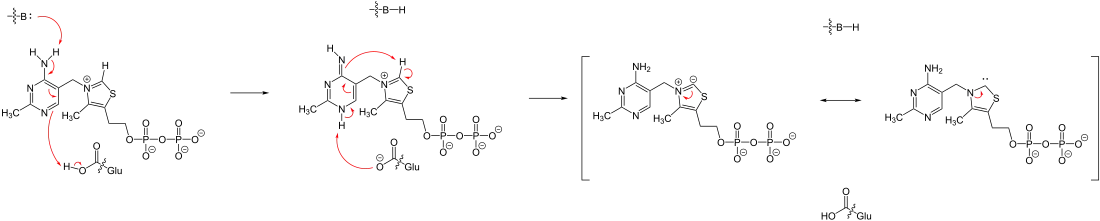
Die negative Ladung wird durch die Resonanzstrukturen 2A und 2B stabilisiert (vgl. Abbildung 3). Das Carben, respektive Carbanion, des Thiazoliumrings greift die Ketogruppe von Pyruvat 1 nucleophil an. Das entstehende Alkoholat 3 wird zum Alkohol 4 protoniert. Die dann folgende Decarboxylierung erfolgt leicht durch den Elektronenzug des Ammoniumions. Die zurückbleibenden Elektronen werden durch Resonanz, 5A und 5B, stabilisiert. Nach Protonierung zu 6 werden der Cofaktor 2A und Acetaldehyd 7 freigesetzt.
Einzelnachweise
- Eintrag bei BRENDA
- John McMurry, Tadhg P. Begley: The organic chemistry of biological pathways. Roberts and Co. Publishers, 2005, ISBN 0-9747077-1-6, S. 179 (englisch).
- PDB 1pyd; F. Dyda, W. Furey, S. Swaminathan, M. Sax, B. Farrenkopf, F. Jordan: Catalytic centers in the thiamin diphosphate dependent enzyme pyruvate decarboxylase at 2.4-A resolution. In: Biochemistry. Vol. 32, Nr. 24, Juni 1993, S. 6165–6170, doi:10.1021/bi00075a008, PMID 8512926 (englisch).
- M. Lobell, D. H. G. Crout: Pyruvate Decarboxylase: A Molecular Modeling Study of Pyruvate Decarboxylation and Acyloin Formation. In: J. Am. Chem. Soc. Vol. 118, Nr. 8, 1996, S. 1867–1873, doi:10.1021/ja951830t (englisch).
- I. Baburina, G. Dikdan, F. Guo, G. I. Tous, B. Root, F. Jordan: Reactivity at the substrate activation site of yeast pyruvate decarboxylase: inhibition by distortion of domain interactions. In: Biochemistry. Vol. 37, Nr. 5, Februar 1998, S. 1245–1255, doi:10.1021/bi9709912, PMID 9477950 (englisch).
- Donald Voet, Judith G. Voet, Charlotte W. Pratt: Lehrbuch der Biochemie. 3. Auflage. Wiley-VCH Verlag, Weinheim 2019, ISBN 978-3-527-34286-0, S. 605.
- Georg Fuchs (Hrsg.): Allgemeine Mikrobiologie. 10. Auflage. Thieme, 2017, ISBN 978-3-13-241885-1, S. 271.
- John McMurry, Tadhg P. Begley: The Organic Chemistry of Biological Pathways. Roberts and Company Publishers, 2005, ISBN 0-9747077-1-6, S. 132 (eingeschränkte Vorschau in der Google-Buchsuche).
- T. D. H. Bugg: Introduction to Enzyme and Coenzyme Chemistry. John Wiley & Sons, 2012, ISBN 978-1-118-34899-4, S. 155 (eingeschränkte Vorschau in der Google-Buchsuche).