Haplotyp
Als Haplotyp (von altgriechisch ἁπλούς haplús, deutsch ‚einfach‘ und τύπος týpos, deutsch ‚Abbild‘, ‚Muster‘), eine Abkürzung von „haploider Genotyp“, wird eine Variante einer Nukleotidsequenz auf ein und demselben Chromosom im Genom eines Lebewesens bezeichnet. Ein bestimmter Haplotyp kann individuen-, populations- oder auch artspezifisch sein.
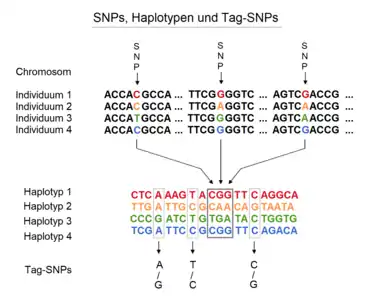
Die dabei verglichenen Allele können, wie beim International HapMap Project, individuelle Kombinationen von SNPs sein, die als genetische Marker benutzt werden können.[1] Besitzt ein Teil der Individuen aufgrund gemeinsamer Abstammung an einem bestimmten Genlocus denselben Haplotyp, werden sie zu einer Haplogruppe zusammengefasst.
Geschichte
Der Begriff wurde 1967 von Ruggero Ceppellini eingeführt.[2] Er wurde ursprünglich dazu benutzt, die genetische Zusammensetzung des MHC zu beschreiben, eines Komplexes von Genen, der für das Immunsystem wichtige Proteine codiert.
Abgrenzung zum Genotyp
Besitzt ein diploider Organismus bezüglich zweier Gene A und B den Genotyp AaBb, so können dem die Haplotypen AB|ab oder Ab|aB zugrunde liegen. Im ersteren Fall besitzt ein Chromosom die Allele A und B, das andere a und b. Im letzteren Fall besitzt ein Chromosom die Allele A und b, das andere a und B.
Bestimmung von Haplotypen
Zwei Fälle können unterschieden werden (im Folgenden bezieht sich der Begriff „Allel“ auf die unterschiedlichen Nukleotide A, C, G und T, jedoch kann z. B. auch die Anzahl der Wiederholung eines bestimmten Mikrosatelliten ein Allel definieren):
- Haploide Spezies
- Die Bestimmung der Haplotypen einer Population haploider Individuen aus derselben Spezies (z. B. verschiedene E. coli Stämme) ist trivial. Hierfür ist die Sequenzierung und Bestimmung der SNPs der gegebenen Population ausreichend (siehe Bild). Werden Individuen bei der Sequenzierung ausgelassen, so können andere darin enthaltene Allele (und die sich daraus ergebenden SNPs) natürlich nicht erfasst werden.
- Polyploide Spezies
- Ist der Ploidiegrad der betrachteten Spezies mindestens 2, so kompliziert sich das Problem (z. B. ist der Mensch diploid, die Kartoffel tetraploid und Weichweizen hexaploid). In diesem Fall wird das Genom aus zwei oder mehr homologen Chromosomensätzen zusammengesetzt, wobei in der Regel die eine Hälfte vom mütterlichen und die andere vom väterlichen Elternteil stammt. Es müssen verschiedene Arten von SNPs unterschieden werden:
- Wenn sich in einem Individuum ein mütterlicher und ein väterlicher homologer Chromosomensatz in Nukleotidpositionen der DNA unterscheiden, so werden diese SNPs bei Sequenzierung der entsprechenden Chromosomen des Individuums sichtbar (es wird immer eine Mischung der homologen Chromosomen sequenziert). Solch ein SNP wird im entsprechenden Individuum heterozygoter SNP genannt.[3]
- Wenn in einem Individuum ein mütterlicher und ein väterlicher homologer Chromosomensatz in einem betrachteten Genlocus identisch ist, so werden bei Sequenzierung der DNA des Individuums keine SNPs sichtbar. Erst wenn bei mindestens einem zweiten Individuum im selben Locus ein anderes Allel gefunden wird, kann an der entsprechenden Nukleotidposition von einem SNP gesprochen werden. Solch ein SNP wird im ersten Individuum homozygoter SNP genannt, kann aber in einem anderen Individuum einen heterozygoten SNP darstellen.[3][4]
- Tauchen in einem SNP zwei verschiedene Allele auf (relativ zur gesamten betrachteten Population), so wird dieser SNP „biallelisch“ genannt. Finden sich drei verschiedene Allele, so wird dieser SNP „triallelisch“ und bei vier Allelen „tetraallelisch“ genannt. Ein tetraallelischer SNP enthält die maximale Anzahl an verschiedenen Allelen, da SNPs nur aus den vier Nukleotiden A,C,G und T gebildet werden können.[4]
- Diploide Spezies können prinzipiell tetraallelische SNPs besitzen, obwohl für ein Individuum nur maximal zwei Allele möglich sind.[4]
Wird nun in einer polyploiden Population (derselben Spezies) ein SNP bestimmt, so lassen sich die Haplotypen (der Länge 1) wie in Punkt 1 direkt aus der Sequenzierung ablesen. Schon bei zwei SNPs wird es problematisch: Bei der Sequenzierung geht die Zuordnung der einzelnen Allele zu ihren ursprünglichen Chromosomen verloren. Verschiedene Kombinationen der Allele in SNP 1 und SNP 2 sind nun möglich und damit auch verschiedene Haplotypen. Die Anzahl der möglichen Haplotypen wächst exponentiell mit der Anzahl der SNPs.
Verschiedene Methoden wurden entwickelt, um Haplotypen in polyploiden Spezies zu bestimmen.
- i) Experimentell:
- Ein gegebenes Chromosom eines gegebenen Individuums wird mehrmals sequenziert und der entsprechende Haplotyp bestimmt. Bei jeder Sequenzierung wurde zufällig eines der homologen Chromosomen aus dem polyploiden Satz ausgewählt. Die Anzahl der Sequenzierungen wird so gewählt, dass mit einer bestimmten Wahrscheinlichkeit davon ausgegangen werden kann, dass kein Haplotyp bei der Sequenzierung ausgelassen wurde.[4] Dies ist teuer und zeitaufwendig. In der Pflanzenzüchtung löst man das Problem durch die Erzeugung ingezüchteter Linien. In letzter Konsequenz sind die homologen Chromosomen eines Individuums aus solch einer Linie reinerbig und demnach identisch (nur homozygote SNPs in einem Individuum). Die Bestimmung der Haplotypen reduziert sich auf Punkt 1 und somit auf eine einmalige Sequenzierung eines Chromosoms bzw. Locus.
- ii) Bioinformatisch:
- Nicht immer sind die Mittel vorhanden, Mehrfachsequenzierungen durchzuführen oder die Möglichkeit gegeben, Inzuchtlinien zu erzeugen. Tauchen nun bei Einmalsequenzierung heterozygote SNPs in einem Individuum auf und wird mehr als ein SNP betrachtet, so können sich verschiedene mögliche Haplotypen für ein Individuum ergeben. Um aus diesen exponentiell vielen Möglichkeiten (bei linear anwachsender Anzahl an SNPs) eine biologisch sinnvolle auszuwählen, wurden verschiedene Methoden, basierend auf unterschiedlichen Annahmen, entwickelt:
- ii.1) Basierend auf einem Sparsamkeitskriterium[5] (Parsimony based, siehe auch Ockham's Razor). Diese Methode versucht, die Anzahl der Haplotypen zu minimieren, welche benötigt werden, um die SNPs einer gegebenen Population zu erklären. Es gibt verschiedene Ansätze basierend auf SAT[3][4] oder Linearer Programmierung[6] dieses Problem effizient zu lösen.
Weitere Eigenschaften: Wird unter der Annahme angewendet, dass im betrachteten Locus keine oder kaum Rekombination stattfindet. Eine gefundene Lösung ist im Sinne des Sparsamkeitskriterium immer optimal. Nicht praktikabel für großmaßstäbliche Analysen. - ii.2) Maximum-likelihood (mit Hilfe von Expectation-Maximization-Algorithmus[7] oder Monte-Carlo-Simulation[8]). Diese Methoden versuchen, den Satz an Haplotypen zu finden (und die entsprechende Aufteilung auf die einzelnen Individuen), so dass die durch eine gegebene Zielfunktion berechnete Wahrscheinlichkeit der beobachteten Daten maximiert wird.
Weitere Eigenschaften: Anwendbar auch bei Rekombination.[9] Lösungen sind meist nur suboptimal, da der Algorithmus in einem lokalen Optimum endet bzw. Vereinfachungen vornehmen muss, damit eine Lösung überhaupt berechenbar ist. Praktikabel für großmaßstäbliche Analysen wenn auch suboptimale Lösung ausreichend ist.
- ii.1) Basierend auf einem Sparsamkeitskriterium[5] (Parsimony based, siehe auch Ockham's Razor). Diese Methode versucht, die Anzahl der Haplotypen zu minimieren, welche benötigt werden, um die SNPs einer gegebenen Population zu erklären. Es gibt verschiedene Ansätze basierend auf SAT[3][4] oder Linearer Programmierung[6] dieses Problem effizient zu lösen.
- Für die Methoden ii.1 und ii.2 ist eine Population mit mehr als einem Individuum notwendig, damit die Grundannahmen greifen und biologisch sinnvolle Aussagen gemacht werden können. Teilprobleme des Haplotypenproblems sind NP-vollständig, da sie durch SAT darstellbar sind (Satz von Cook) und im schlechtesten Fall dieselbe Komplexität wie SAT aufweisen; das Gesamtproblem ist somit NP-schwer.[3]
Nomenklatur der Haplotypen
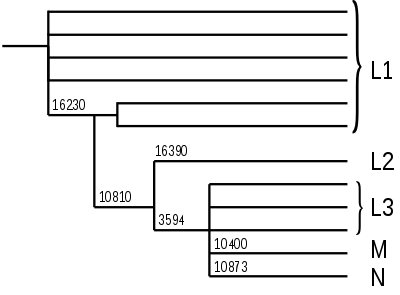
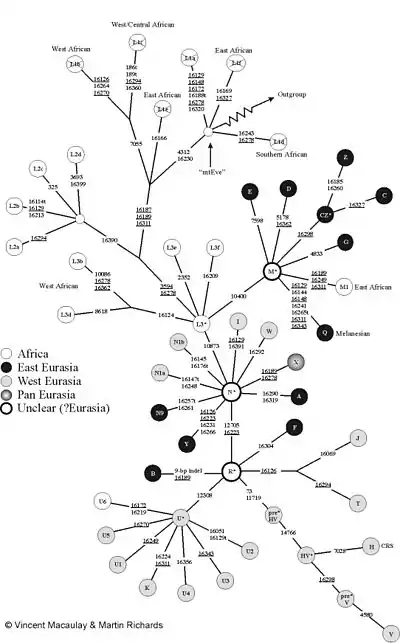
Die Zahlen geben die Position der Mutationen an. „mtEve“ ist die mitochondriale Eva. „Outgroup“ führt zu mtDNA anderer Primaten (z. B. Schimpansen). Die Abbildung benutzt die übliche (falsche) Nomenklatur mit der „L1 Haplogruppe“: L1 bildet jedoch die Wurzel (L1a ist mit L1f nicht näher verwandt als mit V!). Daher wurden die L1-Felder durchgestrichen.[10]
Eine Haplogruppe kann ihrerseits weitere Unter-Haplogruppen enthalten, die sich ihrerseits weiter unterteilen lassen. Man versucht, bei der Nomenklatur der Haplogruppen eine Baumstruktur abzubilden und verwendet abwechselnd Buchstaben und Zahlen. Zwei mtDNAs einer Haplogruppe sind dabei stets monophyletisch. Für die Zuordnung verwendet man charakteristische Mutationen in den Gensequenzen der mtDNA, außerhalb des D-Loops.
| Evolutionsbaum Haplogruppen Mitochondriale DNA (mtDNA) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Eine Person kann z. B. die Haplogruppe C1a3b2 haben. Ihre mtDNA ist dann eng mit der einer anderen Person verwandt, die z. B. C1a3b4 hat. Natürlich teilt ihre mtDNA auch eine gemeinsame Vorfahrin mit einer dritten Person, die C1a3c5 hat, aber diese gemeinsame Vorfahrin hatte früher gelebt, noch bevor sich die C1a3-Linie aufgespalten hatte. Das heißt, C1a3b4 und C1a3b2 sind gegenüber C1a3c5 monophyletisch. Ebenso sind C1a3b2 und C1a3c5 monophyletisch gegenüber allen H-Haplotypen usw.
Die Nomenklatur ist relativ inkonsequent realisiert. Viele Buchstaben wurden benutzt, um die wichtigsten nichtafrikanischen Haplogruppen zu benennen. Viele alte Haplogruppen kommen jedoch in Afrika vor. Diese bezeichnet man zusammen als „L“ und geht bereits für die Unterteilung der Hauptgruppen zu Ziffern über. Über die Zuordnung mancher afrikanischer Haplotypen (in L1 und L3) besteht bis heute noch kein wissenschaftlicher Konsens.
Wenn man von der Wurzel anfängt, besteht der mitochondriale Stammbaum des Menschen zunächst aus einer Reihe tiefer Äste. Diese genetischen Linien werden heute L1 genannt. Anders als früher gedacht, ist L1 keine monophyletische Haplogruppe, sondern bildet die Wurzel. L1 sind also eigentlich ein ganzes Paket afrikanischer Haplogruppen, die ähnlich alt sind wie die mitochondriale Eva und deren genaue verwandtschaftliche Beziehung untereinander noch nicht genau geklärt ist.
Von diesen alten L1-Ästen zweigt ein Ast durch eine Mutation an der Position 10810 ab. Von diesem spaltet sich seinerseits die Haplogruppe L2 durch eine Mutation an der Position 16390 ab. Auch L2 kommt praktisch nur bei Afrikanern südlich der Sahara vor.
Eine Mutation an der Position 3594 bildet den Ast, auf dem die großen Haplogruppen M und N sowie noch zahlreiche weitere afrikanische Haplogruppen, die man heute noch unter L3 zusammenfasst, liegen. L3 ist, wie L1, keine echte (monophyletische) Haplogruppe. Die Haplogruppen M und N kommen beim aller größten Teil der Nichtafrikaner vor. Sie sind in Afrika südlich der Sahara sehr selten, wo L1, L2 und L3 dominieren.
Die Haplogruppe M wird in die großen Haplogruppen M1, Z, C, D, E, G und Q unterteilt. Die Haplogruppe N in N1a, N1b, N9, A, I, W, X und Y, sowie in die Haplogruppe R, die die Unter-Haplogruppen B, F, H, P, T, J, U und K bildet.
Die derzeit umfangreichste Untersuchung von mitochondrialer DNA wurde vom Genographic Consortium durchgeführt (s. a. The Genographic Project). In diesen Vergleich wurden 78.590 genotypische Proben einbezogen und die mitochondrialen Haplogruppen (und deren Untergruppen) wurden in einem phylogenetischen Baum dargestellt.[11]
Geographische Verteilung
Die alten Haplotypen aus den L-Ästen dominieren in Afrika südlich der Sahara. Es bestehen keine Zweifel, dass sie ihren Ursprung dort haben. Diese Haplotypen finden sich auch in Nordafrika (ca. 50 % Häufigkeit) und, in geringer Häufigkeit, in Europa und Westasien.
Die Haplogruppen M und N dominieren im Rest der Welt und sind in Afrika südlich der Sahara selten. Spezielle Varianten der Haplogruppe M (M1) kommen mit einer Häufigkeit von etwa 20 % in Äthiopien vor. Entweder ist M dort bereits entstanden oder es handelt sich um eine semitische Süd-Rückwanderung.
Bei amerikanischen Ureinwohnern kommen die Haplogruppen A, B, C, D und X vor; davon entstanden A, B, und X aus einem Ostzweig der Haplogruppe N, C und D dagegen aus Haplogruppe M.
In Europa und Westasien ist Haplogruppe M extrem selten. Die häufigsten Untergruppen gehören in die Untergruppe R: H, V, T, J, U und K. Daneben kommen auch die Haplogruppen I, W und X mit einer signifikanten Häufigkeit vor. In Europa, dem Kaukasus und dem Nahen Osten finden sich praktisch die gleichen Haplogruppen, nur die Häufigkeiten der einzelnen Haplogruppen schwanken. Vor allem die Haplogruppe H ist im Nahen Osten und im Kaukasus deutlich seltener als in Europa (~25 % versus ~45 %), während die Haplogruppe K deutlich häufiger ist. Innerhalb Europas schwanken die Häufigkeiten der Haplogruppen je nach Region geringfügig.
Süd- und Ostasien unterscheiden sich bei den Haplogruppen sehr stark von Westasien. Hier kommen, aus der Haplogruppe M, die Haplogruppen C, D, E, G, Z und Q vor. Die Haplogruppe N kommt hier auch vor, allerdings ist sie vor allem durch die Haplogruppen A, B, F, Y und X vertreten.
Die Haplogruppe X ist bemerkenswert, da sie in ganz Eurasien und Nordamerika vorkommt, wenn auch nur mit relativ geringer Häufigkeit. Früher wurde angenommen, dass die Haplogruppe X in Europa entstand und nur in Europa vorkommt. Als die Haplogruppe bei amerikanischen Ureinwohnern entdeckt wurde, kam die Hypothese auf, sie sei vor Jahrtausenden von Europa aus auf dem Seeweg durch europäische Emigranten nach Amerika gelangt. Mittlerweile wurde Haplogruppe X jedoch auch in Asien entdeckt (Derneko et al., 2001).
Quellen
Einzelnachweise
- The International HapMap Consortium: The International HapMap Project. In: Nature. Band 426, 2003, S. 789–796 (PDF)
- R. Ceppellini, E. S. Curtoni, P. L. Mattiuz, V. Miggiano, G. Scudeller, A. Serra: Genetics of leucocyte antigens. A family study of segregation and linkage. In: Histocompatibility Testing. 1967, S. 149–185.
- I. Lynce, J. P. Marques-Silva: Efficient haplotype inference with Boolean Satisfiability. In: National Conference on Artificial Intelligence (AAAI). 2006. (PDF).
- J. Neigenfind, G. Gyetvai, R. Basekow, S. Diehl, U. Achenbach, C. Gebhardt, J. Selbig, B. Kersten: Haplotype inference from unphased SNP data in heterozygous polyploids based on SAT. In: BMC Genomics. Band 9, 2008, S. 356 (Zusammenfassung).
- D. Gusfield: Haplotype inference by Pure Parsimony. In: Proceedings of the 14th annual Symposium on Combinatorial Pattern Matching. 2003, S. 144–155. PDF (Memento des Originals vom 10. Juni 2010 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- D. G. Brown, I. M. Harrower: Integer programming approaches to haplotype inference by pure parsimony. In: IEEE/ACM transactions on computational biology and bioinformatics / IEEE, ACM. Band 3, Nummer 2, 2006 Apr-Jun, S. 141–154, ISSN 1545-5963. doi:10.1109/TCBB.2006.24. PMID 17048400.
- L. Excoffier, M. Slatkin: Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. In: Molecular Biology and Evolution. Band 12, 1995, S. 921–927
- Tianhua Niu, Zhaohui S. Qin,4, Xiping Xu, Jun S. Liu: Bayesian Haplotype Inference for Multiple Linked Single-Nucleotide Polymorphisms. In: American Journal of Human Genetics. Band 70, 2002, S. 157–169, PMC 448439 (freier Volltext)
- Shu-Yi Su, Jonathan White, David J. Balding, Lachlan J. M. Coin: Inference of haplotypic phase and missing genotypes in polyploid organisms and variable copy number genomic regions. In: BMC Bioinformatics. Band 9, 2008, S. 513
- Macaulay und Richards
- D.M. Behar u. a.: The Genographic Project public participation mitochondrial DNA database. In: PLoS Genet. Jg. 3, San Francisco 2007, S.e104. PMID 17604454 doi:10.1371/journal.pgen.0030104 ISSN 1553-7390
Literatur
- Lexikon der Biologie. Band 7. Spektrum Akademischer Verlag, Heidelberg 2004, ISBN 3-8274-0332-4.
- Benjamin Lewin: Molekularbiologie der Gene. Spektrum Akademischer Verlag, Heidelberg/Berlin 1998, ISBN 3-8274-0234-4.
Weblinks
- Elke Binder: Jagd nach den Unterschieden. Das internationale „HapMap“-Projekt soll die Suche nach Krankheitsgenen erleichtern. In: Der Tagesspiegel. 26. August 2004 (online [abgerufen am 10. August 2011]).
- Thema und Variation. Ein Katalog der Unterschiede im Erbgut soll die Forschung erleichtern. In: Der Tagesspiegel. 27. Oktober 2005 (online [abgerufen am 10. August 2011]).
- Jan Freudenberg, Sven Cichon, Markus M. Nöthen, Peter Propping: Blockstruktur des menschlichen Genoms: Ein Organisationsprinzip der genetischen Variabilität. In: Deutsches Ärzteblatt. Band 99, Nr. 47, 2002, S. A 3190–3195 (online [abgerufen am 10. August 2011]).