Citrat-Synthase
Die Citrat-Synthase ist dasjenige Enzym (zugehöriges Gen: CS), das die Kondensation von Oxalacetat mit Acetyl-CoA zu Citrat katalysiert. Diese Reaktion ist der erste Schritt im Citratzyklus und daher ist die CS unentbehrlich im Stoffwechsel aller aeroben Lebewesen. Außerdem ist die Reaktion sowie ihre Umkehrung Teil des Citrat-Malat-Pyruvat-Zyklus zur Bereitstellung von NADPH. Die CS ist in den Mitochondrien lokalisiert, das CS-Gen des Menschen befindet sich aber im Zellkern.
| Citrat-Synthase | ||
|---|---|---|
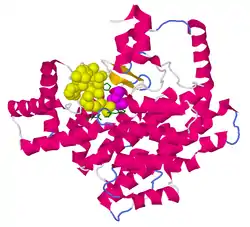 | ||
| Bändermodell des Monomer mit gebundenem Oxalacetat (Magenta) und dem Acetyl-CoA-Analogon CMX (gelb) nach PDB 1CSI | ||
|
Vorhandene Strukturdaten: s. Uniprot | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 439 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Homodimer | |
| Bezeichner | ||
| Gen-Name | CS | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 2.3.3.1, Transferase | |
| Reaktionsart | Übertragung einer Acylgruppe | |
| Substrat | Acetyl-CoA + H2O + Oxalacetat | |
| Produkte | Citrat + CoA | |
| Vorkommen | ||
| Homologie-Familie | Citratsynthase | |
| Übergeordnetes Taxon | Lebewesen | |
Strukturell besteht die CS aus zwei großen Domänen, die fast nur aus α-Helices bestehen (sogenanntes all-α-Protein). Die zwei Domänen sind über ein Zwischenstück verbunden. Es sind zwei Typen der CS bekannt. Während die Citratsynthase in Eukaryoten, grampositiven Bakterien und Archaeen (Typ I) als Dimer vorliegt, bilden die Untereinheiten in gramnegativen Bakterien (Typ II) ein Hexamer (siehe Abb. unten). Zudem ist in Typ II eine der Domänen verglichen mit derselben in Typ I verlängert.[1]
Die katalysierte Reaktion:
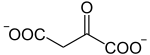 + CH3CO-S-CoA + H2O
+ CH3CO-S-CoA + H2O-
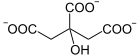 + CoA-SH
+ CoA-SH

Oxalacetat wird mit Acetyl-CoA zu Citrat und CoA umgesetzt, wobei ein Wassermolekül verbraucht wird. Die Reaktion hat eine freie Enthalpie ΔG°’ von −31.5 kJ/mol und ist aufgrund dessen geschwindigkeitsbestimmend für den Citratzyklus.[2]
Die CS ist eines von wenigen Enzymen, die in der Lage sind, ohne die Anwesenheit eines Metallions C-C-Bindungen zu knüpfen.
Reaktionsmechanismus
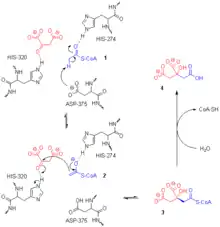
Die Einzelschritte während der Katalyse, die am Dimer doppelt stattfinden, sind:
- Nach der Bindung von Oxalacetat vergrößert sich der Winkel zwischen den zwei α-Domänen um 18°. Diese "Öffnung" des Moleküls versiegelt und schützt einerseits das Oxalacetat, andererseits erlaubt dies erst die zusätzliche Bindung von CoA
- Deprotonierung der Methylgruppe des Acetyl-CoA, ein Enol entsteht und wird stabilisiert
- Nukleophile Addition des Enols an das Carbonyl-C des Oxalacetat, resultierend in Citryl-CoA
- Hydrolyse von Citryl-CoA durch ein deprotoniertes Wassermolekül zu Citrat und CoA-SH
- Rückkehr des Dimers in die Ausgangsstellung
Dieser Mechanismus wurde 1982 von Remington und Kollegen aufgrund kristallografischer Untersuchungen vorgeschlagen.[3]
Bei der Reaktion (Abb. „Mechanismus“) tritt zunächst das Anion des Asparaginsäurerestes ASP-375 als katalysierende Base auf, das, zusammen mit der polarisierend auf die Carbonylgruppe des Acetyl-CoA wirkenden Imidazolyl-Gruppe des Histidinrestes HIS-274, aus der aktivierten Essigsäure 1 über eine Deprotonierung ein als reaktives Zwischenprodukt zu formulierendes Enolat 2 erzeugt, welches analog einer Aldol-Addition mit dem Oxalacetat stereospezifisch zum S-Citryl-CoA 3 reagiert.
Die nachfolgende Hydrolyse des Thioesters zum prochiralen Citrat 4 ist stark exergon (ΔGo < −30 kJ/mol), was diesen Schritt und damit die von der Citrat-Synthase katalysierte Gesamtreaktion irreversibel macht. Citrat als Produkt dieser Reaktion konkurriert mit Oxalacetat um die Bindung an die Citrat-Synthase, so dass eine große Konzentration an Citrat – trotz der hohen Exergonie der Reaktion – die Reaktion inhibiert. Es liegt eine kompetitive Produkthemmung vor.
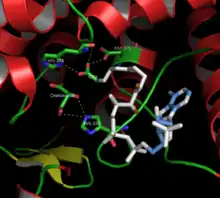
Das in der Abb. wiedergegebene katalytische Zentrum enthält statt des natürlich auftretenden Acetyl-CoA ein synthetisches Analogon (Carboxymethyldethia-CoA, CMX), welches, im Aufbau der Enolform des Acetyl-CoA sehr ähnelnd, gut an dessen Stelle an die Citrat-Synthase bindet, aber nicht als Reaktionspartner im Sinne der Claisen-Kondensation taugt. Dieses Acetyl-CoA-Analoge inhibiert folglich die Reaktion, macht aber die Lage der Substrate im Substrat-Enzym-Komplex sichtbar und ermöglicht somit einen tiefen Einblick in den Mechanismus dieser enzymkatalysierten Reaktion.
Der Mechanismus der Claisen-Kondensation gilt für diese Reaktion mittlerweile als umstritten (auch wenn er weiterhin in den meisten Lehrbüchern anzutreffen ist), da die Abstraktion eines Protons (hoher pKs) durch einen Aspartat-Rest (niedriger pKs) extrem unwahrscheinlich ist. Nach neueren Erkenntnissen bringt das Enzym durch Konformationsänderungen beide Reaktionspartner einander derart nahe, dass das betrachtete Proton weder der Methylgruppe noch dem Aspartat-Rest zugeordnet werden kann und ein Übergang hierdurch begünstigt wird. Dies erinnert an den Mechanismus von Low-barrier hydrogen bonds, welche nur durch räumliche Nähe von Donor und Akzeptor ausgebildet werden können und mit −40 bis −80 kJ/mol deutlich stabiler sind als „gewöhnliche“ Wasserstoffbrücken, doch auch dieses Modell wird dem realen Reaktionsmechanismus nur ansatzweise gerecht. Es muss an dieser Stelle festgestellt werden, dass der Mechanismus der Citrat-Synthase trotz zahlloser Arbeiten und Publikationen auf diesem Gebiet noch immer nicht vollständig geklärt ist.
Weblinks
- Citrate synthase. Proteopedia
Einzelnachweise
- InterPro: IPR019810 Citrate synthase active site (englisch)
- Donald Voet, Judith G. Voet, Charlotte W. Pratt: Fundamentals of Biochemistry: Life at the Molecular Level. Wiley, Hoboken NJ 2008.
- S. Remington, G. Wiegand, R. Huber: Crystallographic refinement and atomic models of two different forms of citrate synthase at 2.7 and 1.7 A resolution. In: Journal of molecular biology, Band 158, Nummer 1, Juni 1982, S. 111–152, ISSN 0022-2836. PMID 7120407.