Adrenogenitales Syndrom
Das adrenogenitale Syndrom (AGS) ist eine Gruppe autosomal-rezessiv vererbter Stoffwechselkrankheiten, die durch eine Störung der Hormonsynthese in der Nebennierenrinde gekennzeichnet sind, wobei es zu einer Überproduktion von androgenen Nebennierensteroiden (Sexualhormone der Nebennierenrinde) kommt. Dabei ist die Bildung von Cortisol und Aldosteron gestört. Durch Überstimulation der Nebennierenrinde werden vermehrt Nebenwege des Stoffwechsels aktiviert und Vorstufen (z. B. Pregnenolon, Progesteron) gebildet. Der Mangel an Cortisol führt zur kompensatorischen Stimulierung der gesamten Nebenniere durch den Hypothalamus und die Hypophyse. Da die Bildung der Sexualhormone – welche dabei nicht gestört ist – ebenfalls in der Nebennierenrinde erfolgt, kommt es bei Mädchen zur Vermännlichung beziehungsweise vorzeitigen Geschlechtsentwicklung beim Jungen. Der Mangel an Aldosteron führt zu Störungen im Salzhaushalt mit Flüssigkeitsverlust. Zur Behandlung müssen die fehlenden Hormone lebenslang ersetzt werden.
| Klassifikation nach ICD-10 | |
|---|---|
| E25.0 | Angeborene Adrenogenitale Störungen in Verbindung mit Enzymmangel |
| ICD-10 online (WHO-Version 2019) | |
Ursache und Einteilung
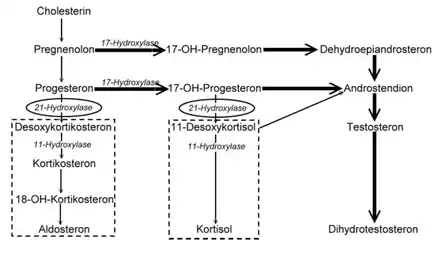
Das adrenogenitale Syndrom wird je nach betroffenem Enzym in fünf Typen unterteilt. Die mit über 90 % der Fälle häufigste Form ist der Typ 3 – auch als Adrenogenitales Salzverlustsyndrom oder Debré-Fibiger-Syndrom bezeichnet – mit Störung des Enzyms 21-Hydroxylase. Es werden je nach Verlauf zwei Formen des 21-Hydroxylase-Mangels unterschieden. Beim klassischen AGS bestehen die Symptome schon im Neugeborenenalter. Es kann mit Salzverlust einhergehen, wenn auch die Aldosteron-Produktion gestört ist. Ist letztere nicht betroffen, besteht kein Salzverlust. Das nicht-klassische AGS manifestiert sich erst später, meist in der Pubertät oder sogar erst im Erwachsenenalter und ist durch eine wesentlich mildere Symptomatik gekennzeichnet.[1][2][3][4][5][6][7]
| Typ | betroffenes Enzym | Genort | Häufigkeit (Geburten) |
| Typ 1 (Lipoid) | StAR-Protein | 8p11.2 | in Europa selten |
| Typ 1 (Lipoid) | Cholesterin-Monooxygenase | 15q23-24 | nur ein Fall bekannt |
| Typ 2 | 3beta-Hydroxysteroid-Dehydrogenase | 1p13.1 | |
| Typ 3 | 21-Hydroxylase | 6p21.3 | 1 : 5.000 – 15.000 |
| Typ 4 | 11-beta-Hydroxylase | 8q24.3 | 1 : 100.000 |
| Typ 5 | 17alpha-Hydroxylase | selten |
Defekte am StAR-Protein treten gehäuft in Japan und Südkorea auf.[8]
Pathophysiologie
Da die Produktion der Nebennierenrindenhormone einem Regelkreis unterliegt, versuchen der Hypothalamus und die Hirnanhangdrüse (Hypophyse) durch vermehrte Ausschüttung des nebennierenrindenstimulierenden Hormons ACTH (adrenocorticotropes Hormon), den Cortisol-Mangel zu kompensieren. Dies führt zu einer Größenzunahme der Nebennierenrinde (Nebennierenhyperplasie) und die Synthesekaskade der Nebennierenrindenhormone wird bis zum Punkt des eigentlichen Enzymdefekts stark angeregt. Zudem kann die erhöhte ACTH-Sekretion auch die Synthese von allen Corticoiden, darunter vor allem Androgenen, deren Sekretion nicht gestört ist, pathologisch direkt stimulieren.[9] Es kommt zu einer vermehrten Bildung der Hormonvorstufen (Steroide), die wegen des Enzymmangels weder zu Cortisol noch Aldosteron, sondern auf alternativen Stoffwechselwegen zu Androgenen abgebaut werden. Die Androgene führen zur Virilisierung.
Der Salzverlust entsteht durch die mangelnde Mineralokortikoid-Wirkung, wenn auch die Synthese von Aldosteron gestört ist. Dies kann auftreten, da die Aldosteron-Synthese in großen Teilen auf identische Enzyme, wie die der Cortisol-Synthese setzt.[9]
Symptome
Die Symptome variieren nicht nur abhängig von der Verlaufsform, sondern unterscheiden sich auch bei den beiden Geschlechtern, da sie zu einem großen Teil durch eine Überproduktion von männlichen Geschlechtshormonen (Androgenen) hervorgerufen werden.
Klassisches adrenogenitales Syndrom ohne Salzverlust
Mädchen, die an dieser Störung erkrankt sind, kommen schon mit einem vermännlichten äußeren Genitale, also einer unterschiedlich stark vergrößerten, fast penisähnlichen Klitoris zur Welt, da sie schon im Mutterleib unter dem Einfluss der vermehrten Androgene standen. Das innere Genitale ist dabei aber weiblich. Jungen haben bei Geburt ein normales äußeres Geschlechtsorgan mit teilweise hyperpigmentiertem Skrotum.[10] Bleibt die Erkrankung unerkannt und dementsprechend unbehandelt, fallen die Kinder durch eine verfrüht einsetzende scheinbare Pubertätsentwicklung (Pseudopubertas praecox) auf. Bereits im Kindesalter können sich eine Achsel- und Schambehaarung entwickeln, der Penis ist stark vergrößert, die Hoden bleiben jedoch kindlich. Bei Mädchen entsteht Hirsutismus. Das Längenwachstum ist zunächst beschleunigt, so dass die Kinder anfangs für ihr Alter zu groß sind. Da es jedoch auch zu einer vorzeitigen Knochenreifung und einem verfrühten Schluss der Wachstumsfugen (Epiphysenfugen) kommt, resultiert daraus schließlich ein Kleinwuchs. Weitere mögliche Symptome sind Akne, mangelnde Brustentwicklung, Störungen des Menstruationszyklus und Unfruchtbarkeit.
Klassisches adrenogenitales Syndrom mit Salzverlust
Bei dieser Verlaufsform bekommen die Kinder zusätzliche Probleme durch die fehlende mineralocorticoide Wirkung des unzureichend gebildeten Aldosteron. Dieses ist maßgeblich für die Wiederaufnahme von Natriumionen aus dem Primärharn durch die Nieren verantwortlich. Betroffene Säuglinge bekommen in den ersten Lebenstagen bis -wochen eine schwere Störung des Salzhaushaltes (Hyponatriämie und Hyperkaliämie) mit Erbrechen und Gewichtsverlust bei metabolischer Übersäuerung des Blutes (Azidose). Dabei werden die Kinder zunehmend teilnahmslos. Diese Salzverlustkrise kann lebensbedrohliche Ausmaße annehmen.
Nicht-klassisches adrenogenitales Syndrom
Diese Verlaufsform, auch als Late-onset-AGS bezeichnet, ist durch milde Symptome eines einfachen AGS ohne Salzverlust gekennzeichnet. Mädchen haben bei Geburt ein normales Genitale. Bei beiden Geschlechtern tritt eine vorzeitige Schambehaarung und Akne auf. Frauen bekommen Störungen des Menstruationszyklus und der Empfängnis. Die Fruchtbarkeit kann beeinträchtigt sein.[11] Der Kleinwuchs macht sich bei beiden Geschlechtern allenfalls geringfügig bemerkbar. Eine Minimalform des AGS, bei dem keine Symptome zu beobachten sind, aber dennoch messbare Veränderungen in den Hormon-Konzentrationen, wird auch nicht-klassisches kryptisches AGS genannt.
Diagnose
Bei Symptomen einer Salzverlustkrise kann eine Bestimmung der Elektrolyte im Serum und des Säure-Base-Status (Blutgasanalyse) Auskunft über die Schwere der Entgleisung geben. Ergibt sich aufgrund der Symptome der Verdacht auf Vorliegen eines Adrenogenitalen Syndroms, kann die Diagnose zunächst durch Bestimmung der Hormonkonzentrationen insbesondere des 17-Hydroxyprogesterons im Blut bestätigt werden. Dieses ist als alternatives Stoffwechselprodukt bei gestörter Kortisol-Synthese deutlich erhöht. In Deutschland gehört die Bestimmung des 17-Hydroxyprogesteron mittlerweile zum erweiterten Neugeborenenscreening und wird bei allen Neugeborenen routinemäßig mitbestimmt. Eine molekulargenetische Untersuchung kann die bekannten zugrundeliegenden Gendefekte nachweisen. Dies ist mittels einer Fruchtwasseruntersuchung (Amniozentese) oder einer Chorionzottenbiopsie auch schon beim Ungeborenen im Mutterleib möglich, so dass betroffene Mädchen auch schon intrauterin durch Korticoid-Substitution behandelt werden können. Um heterozygote Anlagenträger, die selbst nicht erkranken, aber die Veränderung an ihre Kinder weitergeben können, zu identifizieren, kann ein ACTH-Test durchgeführt werden. Dabei wird die Nebenniere durch Gaben des Nebennierenrindenstimulierenden Hormons zu vermehrter Hormonproduktion angeregt. Liegt eine Veränderung auch nur in einem Allel des 21-Hydroxylase-Gens vor, erfolgt ein erhöhter Anstieg von 17-Hydroxyprogesteron.
Therapie
Da die Ursache der Erkrankung in einem Gendefekt liegt, ist eine ursächliche Behandlung nach aktuellen Stand der Wissenschaft nicht möglich. Die symptomatische Therapie besteht in einer lebenslangen Substitution der fehlenden Hormone. Dadurch sinkt die Produktion von ACTH in der Hirnanhangdrüse wieder ab, die Produktion der Androgene nimmt ab, die Nebennierenrinde schrumpft auf normale Größe zurück und die Hormonmangelsymptome verschwinden. Die Therapie erfolgt mit Hydrocortison (bei Erwachsenen oft auch Prednison oder Dexamethason) und kann in Form von Tabletten erfolgen. Da Aldosteron selbst nicht resorbiert wird, erfolgt die Substituierung mit dem Mineralocorticoid Fludrocortison. Mit der Therapie sollte so früh wie möglich begonnen werden. Außerdem muss beachtet werden, dass der Bedarf an Nebennierenrindenhormonen in Stresssituationen deutlich gesteigert ist. Deshalb muss beispielsweise bei akuten Erkrankungen oder vor Operationen die Dosis des Hydrocortisons verdoppelt werden (Fludrocortison wird nicht erhöht). Es gibt keine Indikation die Therapie zu unterbrechen. Alle AGS-Patienten müssen einen Notfallausweis erhalten.
Prognose
Bei guter Einstellung der Hormonsubstitution ist die Prognose des adrenogenitalen Syndroms außerordentlich gut. Die Symptome verschwinden, die Patienten können ein normales Leben führen und erreichen eine normale Fruchtbarkeit.
Siehe auch
- Nebennierenrindeninsuffizienz (Morbus Addison)
- Adrenomyodystrophie
- PCO-Syndrom (als Ausschlussdiagnose nach Diagnose des nicht-klassischen adrenogenitalen Syndroms)
- Pubertas praecox (als Differenzialdiagnose)
Literatur
- Herbert Stolecke (Hrsg.): Endokrinologie des Kindes- und Jugendalters. Springer, Berlin / Heidelberg / New York 1997, ISBN 3-540-61855-4.
Weblinks
Einzelnachweise
- eesom.com prakt. med. Sidonie Achermann, Ärztin
- Adrenogenitales Syndrom. auf: tk.de
- G. Herold: Innere Medizin. Selbstverlag, 2002.
- P. P. Nawroth: Klinische Endokrinologie und Stoffwechsel. Springer, 2001.
- G. Strohmeyer: Angeborene Stoffwechselerkrankungen. Ecomed, 2002.
- D. P. Merke u. a.: Congenital adrenal hyperplasia. In: Lancet. 2005; 365, S. 2125–2136.
- AWMF-Leitlinie Adrenogenitales Syndrom, Leitlinien Register-Nummer 027/047
- OMIM-Eintrag für Typ 1
- Hans-Christian Pape, Armin Kurtz, Stefan Silbernagl: Physiologie. 7. Auflage. Georg Thieme Verlag, Stuttgart 2014, ISBN 978-3-13-796007-2, S. 612, 613.
- Dietrich V. Michalk, Wiebke Ahrens, Eckhard Schönau: Differentialdiagnose Pädiatrie. Elsevier, Urban & Fischer-Verlag, 2005, ISBN 3-437-22530-8, S. 419.
- B. Böttcher, L. Wildt: Gynäkologische Endokrinologie. Diagnose und Therapie. Band 14, Nr. 3, September 2016, S. 212–216, doi:10.1007/s10304-016-0088-9.