Ziliopathie
Eine Ziliopathie ist eine genetisch bedingte Erkrankung der Zilienzellen, deren Grundgerüst (beispielsweise der Basalzellen)[1] oder einer Funktionsstörung der Zilien.[2]
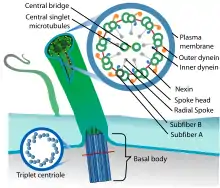
Einführung
Da Zilien auf nahezu allen Zelltypen bei Wirbeltieren vorkommen, wird die elementare Bedeutung dieser Zellorganelle unterstrichen. Es erleichtert das Ausbilden von einem breiten Spektrum an Funktionalität. Die Hauptaufgabe liegt im sensorischen Bereich und einem „Motor für Flüssigkeitsströme“. Im sensorischen Bereich spielen die Zilien eine Rolle bei der Mechanorezeption, dem Geruch, der Lichtwahrnehmung, bei Chemo- und Thermorezeptoren sowie bei der Überwachung des osmotischen Drucks. Die Funktion „Motor für Flüssigkeitsströme“ besteht intra- und extrazellulär, hat Einflüsse z. B. beim Flüssigkeitstransport in der Niere oder den Atemwegen (Flimmerepithel). Daraus lässt sich die Potenz der Beteiligung von Ziliopathien an vielen Krankheiten erahnen. Funktionsstörungen der Zilien machen sich meist schon in der embryonalen Entwicklung bemerkbar. Bei der Stammzellforschung an Zellen von Maus und Mensch wurde die Ausbildung eines primären Ziliums beobachtet. Dieses primäre Zilium ist an elementaren Signalübertragungswegen wie dem Hedgehog (Hh)-, Wnt-, PDGF- und FGF-Signalweg beteiligt. Das Fehlen von Zilien an den sich differenzierenden Zellen scheint eher die Ausnahme zu sein. Das Vorhandensein eines intakten und sich auch differenzierenden Zilienapparates scheint für Zelllinienentwicklung unabdingbar zu sein, da primäre Prozesse der interzellulären Signalübertragungswege davon abhängig sind. Erkenntnisse dazu liegen bisher in Versuchen mit vor allem Mäusen und den bei Aquarianern beliebten Zebrafischen vor. Störungen im Hedgehog-Signalweg führen zu Defekten des Neuralrohrs oder der Fehlentwicklung von Gliedmaßen (Shh-Unterform). Beeinträchtigungen im Wnt-Signalweg führen ebenfalls zum Neuralrohrdefekt und zu Veränderungen an den Stereozilien im Innenohr. Bei weiteren Signalwegstörungen führen Veränderungen am GPCR zur Ausbildung des Laurence-Moon-Biedl-Bardet-Syndroms. Die Ausbildung einer polycystischen Niere steht im Zusammenhang u. a. mit dem zilienabhängingen PDGF-Rezeptor-α. Störungen der Mechanorezeption wurde bei Veränderungen im Bereich der Calcium- und cAMP-gesteuerten Antworten gefunden und führten zu Rechts-Links-Störungen (z. B. Situs inversus) und polycystischen Nieren.[3][4]
Ziliopathien werden überwiegend mit Proteinen assoziiert, bisher wurden ca. 2.500 identifiziert,[5] die den Zilien selber oder Zentrosomen zugeordnet werden. Es ist möglich, dass auch andere Proteine, wie beispielsweise XPNPEP3, das in Mitochondrien lokalisiert ist, vermutlich proteolytische Veränderungen an den Zilienproteinen, und damit Störungen, auslösen.[6]
Erkrankungen
Weitergehende Erkenntnisse dieser Erkrankungsgruppe wurden erst seit Mitte der 1990er Jahre gewonnen, wenngleich viele Funktionen dieser Zellorganellen in den verschiedenen Geweben noch unklar sind. Ein Forschungsschwerpunkt war und ist die Frage, warum Störungen der Zilien so schwere Erkrankungsbilder auslösen können.[7] Da Zilien an nahezu jeder Zelle sitzen und dort ähnliche Aufgaben übernehmen, überlappen sich die Symptome von Erkrankungen häufig und es werden oft übereinstimmende Mutationen an gleichen Genorten gefunden, die Kombination der Mutationen ergibt andere und/oder zusätzliche Syndrombilder
In gesunden Organismen sind besonders „zilienkritische“ Bereiche:
- allgemeine Entwicklung[8]
- Homöostase[8]
- Zeugungsfähigkeit[8]
Bekannte Ziliopathien
| Bezeichnung | **-Nummer | Gen/e | System/Organe |
|---|---|---|---|
| Alström-Syndrom,[1][9] | 203800 | ALMS1 | |
| Laurence-Moon-Biedl-Bardet-Syndrom[9][7][10][11] | 209900 | BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, BBS9, BBS10, TRIM32, BBS12 | |
| Joubert-Syndrom[9][10] | 213300 | INPP5E, TMEM216, AHI1, NPHP1, CEP290, TMEM67, RPGRIP1L, ARL13B, CC2D2A, BRCC3 | Gehirn |
| Meckel-Syndrom[9][10][12] | 249000 | MKS1, TMEM67, TMEM216, CEP290, RPGRIP1L, CC2D2A | Leber, Herz, Knochen |
| Nephronophthisis[9][7][10] | 256100 | NPHP1, INVS, NPHP3, NPHP4, IQCB1, CEP290, GLIS2, RPGRIP1L | Niere |
| Oro-fazio-digitales Syndrom Typ 1[1][7] | 311200 | OFD1 | |
| Senior-Løken-Syndrom[7] | 266900 | NPHP1, NPHP4, IQCB1, CEP290, SDCCAG8 | Auge |
| Zystenniere[9][7] | 173900 | PKD1, PKD2, PKHD1 | Niere |
| Kartagener-Syndrom/Primäre Ciliäre Dyskinesie[9] | 244400 | DNAI1, DNAH5, TXNDC3, DNAH11, DNAI2, KTU, RSPH4A, RSPH9, LRRC50 |
Memo: auf der englischsprachigen Seite der Wikipedia sind alle Links zu den Genorten vorhanden
Andere, gesicherte Ziliopathien
- Fehlgeburt (einige Fälle)[9]
- Hydrocephalus (einige Fälle)[9]
- Kranioektodermale Dysplasie[13]
- Retinopathie (einige Formen)[9]
- Zystenleber[9]
Vermutliche Ziliopathien
Vor allem Badano formuliert in seiner Übersichtsarbeit von 2006 für diese Erkrankungen bzw. partiell für die recht global gehaltenen Symptome eine Ziliopathie als mögliche Ursache:
(Literaturstand bis 2008, bis Ende 2011 keine gesicherte Bestätigung über eine PubMed-Recherche gefunden)
- Corpus-callosum-Agenesie[9]
- Anencephalie[9]
- Störungen der Atmung[9]
- Vermishypoplasie hypoplasia[9]
- Dandy-Walker-Fehlbildung[9]
- Diabetes mellitus[9]
- Ellis-van-Crefeld-Syndrom[9]
- Exencephalie[9]
- Augenbewegungsstörungen[9]
- Lebererkrankungen[9]
- Hypoplasie des Corpus callosum[9]
- Hypotonie[9]
- Unfruchtbarkeit[9]
- Jeune-Syndrom (Asphyxierende Thoraxdysplasie)[9]
- juvenile myoklonische Epilepsie (JME)[14]
- mentale Retardierung/Entwicklungsstörung[9]
- Adipositas[9][10]
- Polydactylie[9]
- posteriore Enzephalozele[9]
- Störungen der Atmungsorgane[9]
- wiederkehrende Atemwegsinfektionen[9]
- Zystenniere[9]
- Retinitis pigmentosa (einige Formen)[9][15] Die Publikation von Khanna suggeriert, dass manche Formen der RP mittlerweile doch eher als bestätigte Ziliopathien anzusehen sind
- Schwerhörigkeit[9]
- Situs inversus[9]
- Spina bifida[9]
- Marden-Walker Syndrom[9]
Einzelnachweise
- M Adams, UM Smith, CV Logan, CA Johnson: Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. In: Journal of Medical Genetics. Band 45, Nr. 5, 2008, S. 257–267, PMID 18178628 (Online [PDF]).
- JH Lee, JG Gleeson: The role of primary cilia in neuronal function. In: Neurobiol. Dis. Band 38, Nr. 2, Mai 2010, S. 167–72, doi:10.1016/j.nbd.2009.12.022, PMID 20097287, PMC 2953617 (freier Volltext).
- JM Gerdes, EE Davis, N Katsanis: The vertebrate primary cilium in Development, Homeostasis, and Disease. In: Cell. Band 137, 2009, S. 32–45, doi:10.1016/j.cell.2009.03.023 (Online).
- C. Edwin, N. Katsanis: Cilia in vertebrate development and disease. In: Development. Band 139, 2012, S. 443–448, doi:10.1242/dev.050054.
- The Ciliary Proteome. Abgerufen am 5. Februar 2012.
- TW Hurd, F Hildebrandt: Mechanisms of Nephronophthisis and Related Ciliopathies. In: Nephron Exp. Nephrol. Band 118, Nr. 1, 2011, S. e9–e14, doi:10.1159/000320888, PMID 21071979, PMC 2992643 (freier Volltext) – (Online).
- JR Davenport, BK Yoder: An incredible decade for the primary cilium: a look at a once-forgotten organelle. In: Am. J. Physiol. Renal Physiol. Band 289, Nr. 6, 2005, S. F1159–69, doi:10.1152/ajprenal.00118.2005, PMID 16275743.
- The Ciliary Proteome. Ciliaproteome V3.0; abgerufen am 15. Juni 2012.
- JL Badano, N Mitsuma, PL Beales, N Katsanis: The ciliopathies: an emerging class of human genetic disorders. In: Annu Rev Genomics Hum Genet. Band 7, 2006, S. 125–48, doi:10.1146/annurev.genom.7.080505.115610, PMID 16722803.
- A Ross, PL Beales, J Hill: The Clinical, Molecular, and Functional Genetics of Bardet-Biedl Syndrome, in Genetics of Obesity Syndromes. Oxford University Press, 2008, ISBN 978-0-19-530016-1, S. 177 (Google Books [abgerufen am 1. Juli 2009]).
- PL Tan, T Barr, PN Inglis u. a.: Loss of Bardet–Biedl syndrome proteins causes defects in peripheral sensory innervation and function. In: Proc. Natl. Acad. Sci. U.S.A. Band 104, Nr. 44, 2007, S. 17524–9, doi:10.1073/pnas.0706618104, PMID 17959775, PMC 2077289 (freier Volltext) – (Online).
- M Kyttälä: Identification of the Meckel Syndrome Gene (MKS1) Exposes a Novel Ciliopathy. (PDF) In: . National Public Health Institute, Helsinki, Mai 2006, archiviert vom Original am 21. Juli 2006; abgerufen am 14. März 2021.
- Dysplasie, kranioektodermale. In: Orphanet (Datenbank für seltene Krankheiten).
- AV Delgado-Escueta: Advances in Genetics of Juvenile Myoclonic Epilepsies. In: Epilepsy Curr. Band 7, Nr. 3, 2007, S. 61–7, doi:10.1111/j.1535-7511.2007.00171.x, PMID 17520076, PMC 1874323 (freier Volltext).
- H Khanna et al.: A common allele in RPGRIP1L is a modifier of retinal degeneration in ciliopathies. In: Nature Genetics. Band 41, 2009, S. 739–745, doi:10.1038/ng.366.