Meckel-Syndrom
Das Meckel-Syndrom (Synonym: Meckel-Gruber-Syndrom, Gruber-Syndrom und Dysencephalia splanchnocystica) ist eine Erbkrankheit. Typisch sind Nierenzysten, die grundsätzlich von weiteren Symptomen wie Leberzysten, Gallengangsdysplasien und Polydaktylie begleitet werden. Abhängig vom zugrunde liegenden Gendefekt werden zahlreiche verschiedene Typen unterschieden. Der Erbgang ist autosomal-rezessiv.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q61.9 | Zystische Nierenkrankheit, nicht näher bezeichnet Meckel-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Als Erstbeschreiber des Syndroms gilt 1822 Johann Friedrich Meckel der Jüngere.[3][4]
Symptome
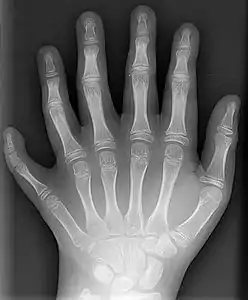
Typisch sind beispielsweise Enzephalozele, Leberzysten, Gallengangsdysplasien, Zystennieren, Polydaktylie, Augenanomalien, Lippen-Kiefer-Gaumen-Spalte, Leberfibrose, Spina bifida und Herzfehler. Um die Diagnose „Meckel-Syndrom“ stellen zu können, müssen mindestens zwei typische Veränderungen vorhanden sein. Die noch im ICD10-Code als „zwingend vorhanden“ abgebildete polyzystische Nierendegeneration gilt heutzutage nicht mehr als notwendiges Kriterium.[4]
Verbreitung
Die Häufigkeit wird mit 1 zu 38.500 Geburten in Europa angegeben,[2] seine Häufigkeit regional unterschiedlich. Gehäuft tritt es in Finnland auf (Inzidenz 1:140.000–9000).[4]
Ursache
Je nach zugrunde liegender Mutation können folgende Typen unterschieden werden:
- Typ 1 (finnischer Typ): MKS1-Gen auf Chromosom 17 Genort q23[5]
- Typ 2 (arabischer Typ): TMEM216-Gen auf Chromosom 11 an q12.2[6]
- Typ 3 (indischer Typ): TMEM67-Gen auf Chromosom 8 an q22.1[7]
- Typ 4: CEP290-Gen auf Chromosom 12 an q21.32[8]
- Typ 5: RPGRIP1L-Gen auf Chromosom 16 an q12.2[9]
- Typ 6: CC2D2A-Gen auf Chromosom 4 an p15.32[10]
- Typ 8: TCTN2-Gen auf Chromosom 12 an q24.31[11]
- Typ 9: B9D1-Gen auf Chromosom 17 an p11.2[12]
- Typ 11: TMEM231-Gen auf Chromosom 16 an q23.1[13]
- Typ 13: TMEM107-Gen auf Chromosom 13 an p13.1[14]
Mutationen in diesem Gen finden sich auch bei einer Form des Joubert-Syndroms.
Das Meckel-Syndrom Typ 7 wird als NPHP3-assoziiertes Meckel-ähnliches Syndrom, Synonym: Meckel-ähnliches-Syndrom Typ 1 geführt.[15]
Therapie und Prognose
Die Prognose ist abhängig von der Art und dem Grad der Ausprägung der vorhandenen Fehlbildungen, aber grundsätzlich als infaust zu betrachten. Neugeborene überleben meist nur wenige Tage.[16] Eine spezifische Therapie ist nicht möglich, in manchen Fällen sind lindernde Maßnahmen angezeigt.[4]
Einzelnachweise
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Meckel-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- L. Göbbel: Pathomorphologische Untersuchungen und Screening von aDNA-Sequenzen zur Detektion von Aneuploidien in humanteratologischen Präparaten der Meckel’schen Sammlungen. 2008, S. 54, Abstract und 8. Thesen. (PDF; 56 kB); abgerufen am 23. April 2009.
- R. Witkowski u. a.: Lexikon der Syndrome und Fehlbildungen. Springer, 2003, ISBN 3-540-44305-3, S. 801; books.google.de
- Meckel syndrome 1. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 2. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 3. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 4. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 5. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 6. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 8. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 9. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 11. In: Online Mendelian Inheritance in Man. (englisch)
- Meckel syndrome 13. In: Online Mendelian Inheritance in Man. (englisch)
- NPHP3-assoziiertes Meckel-ähnliches Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- A. Strauss: Meckel-Gruber-Syndrom. In: Ultraschallpraxis. 2. Auflage. 2008, ISBN 978-3-540-78252-0, S. 293. springer.com