Asphyxierende Thoraxdysplasie
Die Asphyxierende Thoraxdysplasie ist eine sehr seltene angeborene Skelettdysplasie aus der Gruppe der Kurzripp-Polydaktylie-Syndrome mit sehr kurzen Rippen und entsprechend schmalem Thorax, kurzen Gliedmaßen und typischen Veränderungen im Röntgenbild. Sie zählt zu den Ziliopathien und verläuft häufig letal.[1][2][3]
| Klassifikation nach ICD-10 | |
|---|---|
| Q77.2 | Kurzripp-Polydaktylie-Syndrome Inkl.: Asphyxierende Thoraxdysplasie [Jeune] |
| ICD-10 online (WHO-Version 2019) | |
Synonyme: Asphyxierende Thoraxdystrophie, lateinisch Thorako-pelviko-phalangeale Dystrophie; Jeune-Syndrom; englisch Asphyxiating thoraxic dystrophy; ATD; Thoracic-pelvic-phalangeal dystrophy; Short-rib thoracic dysplasia; SRTD
Die Namensbezeichnung bezieht sich auf den Erstautor der Erstbeschreibung aus dem Jahre 1954 durch den französischen Kinderarzt Mathis Jeune und Mitarbeiter.[4]
Vorkommen
Die Häufigkeit wird auf 1–5 auf 500 000 geschätzt. Die Krankheit wird autosomal-rezessiv vererbt.[1]
Ursache
Je nach zugrunde liegender Mutation können folgende Typen unterschieden werden:
- Typ 1 mit Mutationen auf Chromosom 15 Genort q13[5]
- Typ 2 mit Mutationen im IFT80-Gen auf Chromosom 3 Genort q25.33[6]
- Typ 3 (Verma-Naumoff-Syndrom) mit Mutationen im DYNC2H1-Gen auf Chromosom 11 Genort q22.3[7]
- Typ 4 mit Mutationen im TTC21B-Gen auf Chromosom 2 Genort q24.3[8]
- Typ 5 mit Mutationen im WDR19-Gen auf Chromosom 4 Genort p14[9]
- Typ 10 mit Mutationen im IFT172-Gen auf Chromosom 2 Genort p23.3[10]
- Typ 11 mit Mutationen im WDR34-Gen auf Chromosom 9 Genort q34.11[11]
Jedes dieser Gene kodiert für ein Flagellen-Transportprotein, eventuell gibt es noch weitere, bislang unbekannte Mutationen.[1]
Klinische Erscheinungen
Die Erkrankung kann bereits intrauterin erfasst werden[12], fällt ansonsten unmittelbar nach der Geburt auf.
Der Ausprägungsgrad der Veränderungen kann beträchtlich unterschiedlich sein. Klinische Kriterien sind:[1][2] Thoraxdeformität mit langem, schmalen Thorax mit Atemnot des Neugeborenen infolge Platzmangels für die Lunge; Kurze Extremitäten mit Kleinwuchs, eventuell Polydaktylie; Im späteren Kindesalter können Niereninsuffizienz, Retinopathie, Fibrosierung von Leber und Pankreas auftreten.
Diagnose
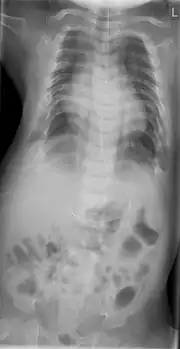
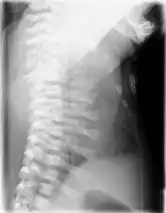
Die klinische Verdachtsdiagnose wird im Röntgenbild gesichert. Diagnostische Kriterien sind:[1][2][13]
- Kurze, horizontal verlaufende Rippen
- Schmale Beckenschaufeln mit dreizackartiger Begrenzung des Unterrandes, Pfannendach horizontal verlaufend und spornartig nach kaudal weisend
- Vorzeitige Verknöcherung der Femurepiphyse
- Distal betont verkürzte Röhrenknochen
- Zapfenepiphysen der Mittelphalangen
Formen
Nach der Art der Verknöcherung der Knorpelanlagen können zwei Formen unterschieden werden:[14]
- Typ I Fleckförmige enchondrale Ossifikation, unregelmäßige Metaphysen, Knorpelinseln in den Metaphysen
- Typ II regelmäßige Ossifikation
Differential-Diagnose
- Thanatophore Dysplasie
- Metatrophischer Dwarfismus
- Hypophosphatasie
- Ellis-van-Creveld-Syndrom, die Abgrenzung kann sehr schwierig sein.[15]
- Thorax-Kehlkopf-Becken-Dysplasie, Synonyme: Barnes-Syndrom; Thorax-Larynx-Becken-Dysplasie; Thorakopelvine Dysostose[16]
- Kranioektodermale Dysplasie (Sensenbrenner-Syndrom)
- paternale uniparenterale Disomie des Chromosoms 14
Therapie
Die Behandlung und deren Erfolg hängen von dem Ausmaß der Lungenbeeinträchtigung ab, auftretende Infektionen der oberen Luftwege müssen behandelt, Nieren-, Leberfunktion und Augenhintergrund regelmäßig kontrolliert werden.
Überleben die Kinder erholt sich die Lungenfunktion allmählich, das Risiko für Atemwegsinfekte nimmt dann ab.[1]
Eine neuere Behandlungsmöglichkeit ist durch die Vertical expandable prosthetic titanium Ribs (VEPTR) nach Campbell[17][18] entstanden, bei welcher der Rippenthorax operativ gedehnt werden kann.
Literatur
- R. S. Drebov, A. Katsarov, E. Gagov, N. Atanasova, Z. Penev, A. Iliev: Is Asphyxiating Thoracic Dystrophy (Jeune's Syndrome) Deadly and Should We Insist on Treating It? Reconstructive Surgery "On Demand". In: Surgery journal. Band 3, Nummer 1, Januar 2017, S. e17–e22, doi:10.1055/s-0037-1598043, PMID 28825014, PMC 5553486 (freier Volltext).
- M. Schmidts, Y. Hou, C. R. Cortés et al.: Corrigendum: TCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport. In: Nature Communications. Band 7, März 2016, S. 11270, doi:10.1038/ncomms11270, PMID 27021811, PMC 4820607 (freier Volltext).
- R. Shaheen, M. Schmidts, E. Faqeih et al: A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. In: Human Molecular Genetics. Band 24, Nummer 5, März 2015, S. 1410–1419, doi:10.1093/hmg/ddu555, PMID 25361962, PMC 4321448 (freier Volltext).
- D. Saletti, T. R. Grigio, D. Tonelli, O. D. Ribeiro Júnior, F. Marini: Case report: anesthesia in patients with asphyxiating thoracic dystrophy: Jeune syndrome. In: Revista brasileira de anestesiologia. Band 62, Nummer 3, 2012 May-Jun, S. 424–431, doi:10.1016/S0034-7094(12)70142-3, PMID 22656687.
- M. Jeune, C. Beraud, R. Carron: Dysthrophie thoracique asphyxiante de caractère familial. In: Archives françaises de pédiatrie. Band 12, Nummer 8, 1955, S. 886–891, ISSN 0003-9764. PMID 13292988.
- J. de Vries, J. L. Yntema, C. E. van Die, N. Crama, E. A. Cornelissen, B. C. Hamel: Jeune syndrome: description of 13 cases and a proposal for follow-up protocol. In: European Journal of Pediatrics. Band 169, Nummer 1, Januar 2010, S. 77–88, ISSN 1432-1076. doi:10.1007/s00431-009-0991-3. PMID 19430947. PMC 2776156 (freier Volltext).
Einzelnachweise
- Jeune-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- emedicine.medscape.com
- M. Jeune, R. Carron, C. Beraud, Y. Loaec: Polychondrodystrophie avec blocage thoracique d’évolution fatale. In: Pédiatrie. Band 9, Nummer 4, 1954, S. 390–392, ISSN 0031-4021. PMID 13185703.
- Short-rib thoracic dysplasia 1 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 2 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 3 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 4 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 5 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 10 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- Short-rib thoracic dysplasia 11 with or without polydactyly. In: Online Mendelian Inheritance in Man. (englisch)
- D. W. Bianchi, T. M. Crombleholme, M. E. D’Alton: Fetology: diagnosis & management of the fetal patient. 2000 In: McGraw-Hill Professional, ISBN 978-0-8385-2570-8, S. 673–
- J. W. Spranger: Bone Dysplasias, Urban & Fischer 2002, ISBN 3-437-21430-6.
- F. Hefti: Kinderorthopädie in der Praxis. Springer Berlin u. a. 1997, ISBN 978-3-540-61480-7
- L. Langer: Thoracic-pelvic phalangeal dystrophy 1968 in: Radiology 91, S. 447.
- Dysostose, thorakopelvine. In: Orphanet (Datenbank für seltene Krankheiten).
- A. K. Hell, R. M. Campbell, F. Hefti: Neues Therapiekonzept für Kinder mit Thoraxinsuffizienz-Syndrom aufgrund von Wirbelsäulenfehlbildungen In: Klinische Pädiatrie. Band 217, Nummer 5, 2005 Sep-Oct, S. 268–273, ISSN 0300-8630. doi:10.1055/s-2004-832483. PMID 16167273.
- M. Lacher, H. G. Dietz: VEPTR (Vertical Expandible Prosthetic Titanium Rib) treatment for Jeune syndrome. In: European journal of pediatric surgery : official journal of Austrian Association of Pediatric Surgery ... [et al] = Zeitschrift für Kinderchirurgie. Band 21, Nummer 2, März 2011, S. 138–139, ISSN 1439-359X. doi:10.1055/s-0030-1267222. PMID 21053163.