Morbus Hippel-Lindau
Das Von-Hippel-Lindau-Syndrom (VHL-Syndrom), genannt auch Hippel-Lindausche Krankheit, gelegentlich auch als Retino-cerebelläre Angiomatose bezeichnet, ist eine seltene, erbliche Tumorerkrankung aus dem Formenkreis der sogenannten Phakomatosen.[1] Die Patienten entwickeln gutartige, geschwulstähnliche Gewebsveränderungen (Angiome) vornehmlich im Bereich der Netzhaut des Auges und des Kleinhirns. Letzteres wurde auch als Lindau-Tumor oder Lindau-Syndrom bezeichnet. Im zentralen Nervensystem können darüber hinaus auch der Hirnstamm und das Rückenmark, selten das Großhirn betroffen sein. Charakteristisch für das VHL-Syndrom ist, dass sich aus Vorformen des Bindegewebes Geschwülste entwickeln, die aus Gefäßknäueln bestehen. Viele Patienten haben auch Gewebsveränderungen im Bereich von Niere (Nierenzellkarzinome), Nebenniere (Phäochromozytom) und Bauchspeicheldrüse. Bei Männern kann der Nebenhoden betroffen sein. Diese Gewebsveränderungen können harmlos sein, sich aber auch zu bösartigen Tumoren entwickeln. Die Ursache der Erkrankung ist eine Genmutation im Von-Hippel-Lindau-Tumorsuppressor-Gen. Da es sich bei dem VHL-Syndrom um eine genetische Erkrankung handelt, ist keine Heilung möglich. Die Behandlung der Patienten richtet sich nach dem Ort und der Ausprägung der Gewebsveränderungen. Am Auge werden die Netzhautgeschwülste mittels Laserstrahlen zerstört. Bösartige Geschwülste treten vor allem im Bereich der Niere auf und werden gemäß den Richtlinien der Behandlung dieser Erkrankung behandelt. Die Gewebsveränderungen im zentralen Nervensystem werden operiert, wenn durch deren Lage und Größe gefährliche Folgen für die Patienten eintreten können. Da die Krankheit schon frühzeitig erkannt werden kann, werden regelmäßige Kontrolluntersuchungen empfohlen.
| Klassifikation nach ICD-10 | |
|---|---|
| Q85.8 | Sonstige Phakomatosen |
| ICD-10 online (WHO-Version 2019) | |
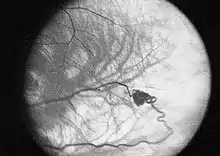
Geschichte
Namensgeber der Erkrankung sind der Göttinger Ophthalmologe Eugen von Hippel (1867–1939) und der schwedische Pathologe Arvid Lindau (1892–1958). Von Hippel beschrieb 1904 erstmals Angiome des Auges, Lindau 1926 die Angiome im Rückenmark.
Inzidenz, Erbgang, Epidemiologie
Ein familiärer Zusammenhang der Erkrankung wurde zunächst 1894 bei der Untersuchung von Geschwistern vermutet.[2] 1929 wurde festgestellt, dass das VHL-Syndrom einer autosomal-dominanten Vererbung gehorcht.[3] Mit zunehmendem Alter steigt das Risiko Tumoren zu bilden. Dabei wird das Maximum der genetischen Penetranz im Alter von 65 Jahren erreicht.[4] Die Inzidenz wird zwischen 1:36.000 - 1:45.000 angegeben.[5] Die möglichen Ausprägungen der Erkrankung können bei Erkrankten stark variieren.[6] Je nach Studie beträgt die Spontanmutationsrate bis zu 50 %. Männer wie Frauen sind gleichermaßen betroffen.
Pathogenese, Molekularbiologie und pathophysiologische Zusammenhänge
Das Gen für die Hippel-Lindau Erkrankung wurde im Bereich von Chromosom 3, Bande p25/26 lokalisiert. Es ist im Zellzyklus und der Gefäßneubildung involviert. Das HL-Gen besitzt drei Exons und kodiert für ein nukleäres Protein, das eine Bindung mit Proteinen der Elongin-Gruppe eingeht. Bei Patienten mit einer HL-Erkrankung wurde eine Vielzahl von Mutationen entdeckt, die alle weitgehend gleichmäßig über das Gen verteilt sind. In verschiedenen Studien wurde festgestellt, dass 35 % der Mutationen Missense-Mutationen sind und etwa 75 % der Patienten haben eine Keimbahnmutation.
Pathologie
Bei den Angiomen des HL-Syndroms handelt es sich vorwiegend um kapilläre Hämangiome und Hämangioblastome. Als Hämangiom bezeichnet man gutartige, geschwulstähnliche Neubildungen mit einer Ausprägung als Blutgefäßknäuel. Sie treten meist als Hamartome auf, sind also keine Tumoren im engeren Sinne. Die Hämangiome entstehen nicht, wie der Name nahelegt, aus Blutgefäßen, sondern aus Bindegewebsvorläufern und sie entwickeln sich zu Strukturen, die man am einfachsten als Blutgefäßknäuel oder Blutschwämme bezeichnen kann. Die Blutschwämme, die sich im Kleinhirn, Hirnstamm und Rückenmark der VHL-Patienten finden, sind Hämangioblastome. Mit diesem Begriff bezeichnet man echte Neubildungen, die aus gewucherten Kapillarsprossen bestehen.
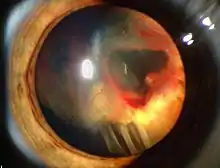
- Die retinalen Hämangiome können eine mehr angiomatöse oder mehr fibrosierende Ausprägung haben. Dies ist für die Prognose der Erkrankung in Bezug auf das Auge sehr wichtig. Bei mehr angiomatösen Hämangiomen überwiegt der Gefäßanteil der Gewebsveränderung, was häufiger zu massiven Einblutungen in das Auge mit einer plötzlichen vollständigen Erblindung führen kann. Fibrosierende Hämangiome machen eher eine Traktionsveränderung der Netzhaut. Wenn die Hämangiome in der Peripherie der Netzhaut gelegen sind, machen sie häufig keinerlei Beschwerden. Liegen sie dagegen zentral, kann bald eine Visusminderung auftreten. Wenn die Gefäßknäuel Kurzschlüsse zwischen Venen und Arterien ausbilden, kann es zum Austreten von Gewebsflüssigkeit im Auge kommen mit entsprechenden Folgen (Druckerhöhung).
- Hämangioblastome manifestieren sich bevorzugt im Kleinhirn und werden bei langsamem Wachstum durch Störung des Liquorabflusses symptomatisch. Da sie auch Erythropoetin ausbilden, wird in manchen Fällen eine Polyzythämie beobachtet. Unter dem Mikroskop stellt sich das kapilläre Hämangioblastom als äußerst gut vaskularisierter Tumor dar mit CD31/CD34-positivem Gefäßendothel und NSE-Expression des Stromas dar.
Krankheitsbild
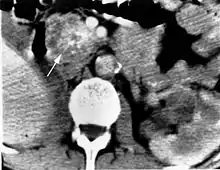
Die Kern- oder Kardinalsymptome des VHL-Syndroms sind das Auftreten von Hämangiomen der Retina und Hämangioblastomen des Kleinhirns.
Das klinische Spektrum der Erkrankung umfasst neben den Affektionen von Augen und Kleinhirn das Auftreten von Hämangioblastomen im Bereich des Hirnstamms und des Rückenmarkes. Sodann werden Nierenzellkarzinome (Erkrankungsrisiko liegt bei 25 – 45 %, meist ab dem 50. Lebensjahr), Pankreaszysten, Phäochromozytome, Nebenhodenzysten und eine Polyzythämie beobachtet.
Die Hämangioblastome des ZNS präsentieren sich in ca. 60 % der Fälle im Kleinhirn, in 13 % der Fälle im Rückenmark und in 4 % der Fälle im Hirnstamm. Selten < 1 % im Großhirn. Die Läsionen im Kleinhirn werden im Schnitt bei Patienten im Alter von 29 Jahren klinisch auffällig und im Fall von Rückenmarksläsionen im Alter von 34 Jahren.
Als diagnostisches Kriterium gilt der Nachweis von beidseitigen oder multiplen retinalen Hämangiomen oder der Nachweis multipler Hämangioblastome der hinteren Schädelgrube.
Aufgrund klinischer Verläufe und Unterschieden in der Phänotyp-Genotyp-Korrelation unterscheidet man zwei Formen des VHL-Syndroms. Patienten ohne Phäochromozytom ordnet man dem VHL-Typ I zu, Patienten mit einem Phäochromozytom dem VHL Typ II.
Zum Verlauf der Erkrankung werden sehr unterschiedliche Angaben gemacht. Die retinalen Angiome werden im Durchschnitt bei den Patienten im Alter von 25 aufgedeckt. Die Netzhautveränderungen können, sofern sie rechtzeitig entdeckt werden gut behandelt werden. Die intrazerebralen und spinalen Hämangioblastome können vor allem im Bereich des Hirnstammes zu gefährlichen Blutungen führen.
Genotyp-Phänotyp-Korrelation
Bei dem HL-Typ I wurden in über 50 % der untersuchten Fälle Mikrodeletionen und nonsense-Mutationen entdeckt. Beim HL-Typ II fanden sich in fast 100 % der untersuchten Patienten missense-Mutationen.
Diagnose
Die Diagnose VHL-Syndrom wird beim Vorhandensein von kapillären Hämangioblastomen (gefäßreichen Tumoren) im ZNS oder der Netzhaut des Auges gestellt. Weitere zum VHL-Komplex gehörende Tumoren (Phäochromozytom, Nierenzellkarzinom) oder eine entsprechende Familienanamnese treten hinzu. In der Kernspintomografie stellen sich die Hämangioblastome als kontrastmittelaufnehmende Knötchen dar.
Therapie
Die Wahl der Therapie ist von der Größe, Lokalisation und vom klinischen Bild abhängig. Nur eine frühzeitige Diagnose und effektive Therapie kann eine Erhaltung des Visus ermöglichen.[7] In einigen seltenen Einzelfällen wurden spontane Regressionen ohne Therapie der retinalen Angiomen beschrieben.[8][9]
Laserkoagulation
Die Laserkoagulation wird zurzeit bei kleineren retinalen Hämangiomen angewendet. Dabei werden Argon-,[10][11] Krypton-,[12] Farbstoff- und Diodenlaser, früher auch Xenon-Koagulator, eingesetzt. Der Vorteil des Verfahrens ist die gezielte Zerstörung mit genauer Dosierung, wobei das gesunde Gewebe geschont wird.[13] Durch häufige Anwendungen der Methode zeigen sich Erfolge bei retinalen Hämangiomen bis zu einer Größe von 4,5 mm, allerdings ist die Therapie am effektivsten bei Größen bis 1,5 mm (entspricht einem Papillendurchmesser) oder kleiner.[14][15][16] Angiome, die größer als ein Papillendurchmesser sind, zeigen bei der Laserkoagulation nur unbefriedigende Ergebnisse und sollten daher mit anderen Methoden behandelt werden.[17] Die Laserkoagulation kann direkt auf das Angiom erfolgen, auf die zuführende Gefäße des Angioms oder beides gleichzeitig.[18] Die Ansprechrate bei der direkten Photokoagulation bei der Benutzung vom Argon-Lasor liegt bei 91 – 100 %.[15][19] Hämangiome mit der Größe von einem Papillendurchmesser brauchen durchschnittlich drei Anwendungen bis zur vollständigen Verödung, hingegen ist bei Mikroangiomen eine Sitzung meist ausreicht.[17] Einige Autoren empfehlen die Kombination der Laserkoagulation mit anderen Methoden, um Angiome zu behandeln, die durch die alleinige Laserkoagulation nicht ausreichend behandelt werden konnten.[20]
Die Verwendung des gelben Lasers beim Krypton-Laser bietet einen theoretischen Vorteil gegenüber den anderen Laserverfahren, da das oxidierte und reduzierte Hämoglobin mehr gelbe Wellenlängen absorbiert, als grüne oder blaue wie beim Argon-Laser.[21] Dadurch sollten die zuführenden Gefäße stärker bestrahlt werden.
Als häufigste Nebenwirkungen der Laserkoagulation können Glaskörperblutungen oder exsudative Netzhautablösungen, speziell durch Lipidablagerungen in der Makula mit bleibendem Visusverlust, auftreten.[14][22] Dieser degenerative Vorgang kann innerhalb von einem Tag nach der Behandlung auftreten.[23] Die Komplikationen treten besonders häufig auf, wenn das Angiom schon strukturelle Veränderungen verursacht hat.[13] Bei der indirekten Laserkoagulation kann es, nach einigen Monaten nach der Behandlung, zu einer Reperfusion der zuführenden Gefäße des Angioms kommen.[24] Wodurch Nachbehandlungen notwendig werden können. Der Verschluss einer zuführenden Arterie kann zu einem Infarkt der von ihr versorgenden Netzhaut nach sich ziehen. Die Folge sind Gesichtsfeldausfälle.[11]
Trotz einer Behandlung mit der direkten oder indirekten Laserkoagulation können sich später neue Hämangiome entwickeln.[25][17] Auch Rezidive des behandelten Angioms können auftreten.[26] In histologischen Untersuchungen zeigten sich bei der Behandlung mit Laserstrahlen nur eine oberflächliche Zerstörung von Tumorzellen bei großen Hämangiomen. In der Tiefe konnte unverändertes Tumorgewebe gefunden werden. Im Gegensatz dazu konnte eine vollständige Zerstörung bei kleinen Hämangiomen nachgewiesen werden.[24]
Kryotherapie
Bei der Kryotherapie werden Angiome, die größer als zwei Papillendurchmesser sind und weit peripher liegen, behandelt. Auch Angiome mit subretinalen Exsudaten können so behandelt werden.[17][27] Diese Methode wurde 1967 zum ersten Mal von Lincoff eingesetzt und untersucht.[28] Dabei wird das retinale Angiom bei Temperaturen von −60 °C bis −80 °C vereist.[29] In einer Langzeitstudie zeigten sich die besten Ergebnisse bei Angiomen, die kleiner als 3,75 mm waren. Bei größeren Angiomen war die Kryotherapie nicht erfolgreicher als die Laserkoagulation, zeigte aber weniger Komplikationen als mit der Behandlung mit Laserstrahlen.[19]
Auch bei der Kryotherapie sind häufig Nachbehandlungen notwendig, allerdings sollte ein Mindestabstand von zwei Monaten zwischen den Behandlungen eingehalten werden.[30] Als Komplikationen können exsudative Netzhautablösungen und eine Proliferative Vitreoretinopathie auftreten.[7]
Brachytherapie
Bei Angiomen von einer Größe von 4 mm können Ruthenium-106 Applikatoren als episkeral aufgenähte Stahlträger eingesetzt werden. Nach der Bestrahlung kommt es, im Vergleich zur Behandlung mit Laserstrahlen, zu einer langsameren Zurückbildung der Angiome. Die durchschnittliche Regressionsdauer beträgt 5 bis 14 Monate.[31][32][33] Nach der Bestrahlung entstehen fast immer chorioretinale Narben, die größer als der Tumor waren. Als größte Gefahr besteht die Verletzung des Sehnervs durch die radioaktiven Strahlen.
Auch bei der Brachytherapie können exsudative Netzhautablösungen und Glaskörperblutungen auftreten, die jedoch seltener auftraten als bei den herkömmlichen Behandlungsmethoden. Selten können epiretinale Membranbildungen auftreten, die eine chirurgische Sanierung notwendig macht. Bei einer, vor der Behandlung bestehenden, Netzhautablösung ist das Risiko für Komplikationen nach der Brachytherapie jedoch stark erhöht.[31]
Transpupilläre Thermotherapie
Die Transpupilläre Thermotherapie kann bei der Behandlung von chorioidalen Melanomen[34], Retinoblastom[35] und chorioidale Hämangiome[36] versucht werden. Dabei wird mittels Infrarot-Diodenlaser eine lokale Erwärmung beim Tumor erzeugt.[37] Histologische Untersuchungen zeigten nach der Behandlung deutliche Nekrosen des Tumors beim chorioidalen Melanom.[38] Bei der Behandlung von retinalen Hämangiomen zeigten sich einerseits Erfolge,[39] andrerseits zeigten andere Studien kaum Wirksamkeit der Methode.[40]
Photodynamische Therapie
Bei größeren Angiomen wird die Effektivität der photodynamischen Therapie mit Verteporfin in klinischen Studien zurzeit untersucht.[41] Dabei konnten Verbesserungen des Visus um durchschnittlich 0,5 nach zwei Jahren festgestellt werden. Als Nebenwirkung kann ein Makulaödem auftreten, welches wiederum zur Visusverschlechterung führen kann.[42][43] Diese Methode kann auch mit der Photokoagulation kombiniert werden.[44]
Strahlentherapie
Die Behandlung mit Strahlen wurde als aller erste Therapiemethode bei retinalen Angiomen durch Houwer 1919 versucht.[45] Dabei konnte er jedoch keine Wirksamkeit feststellen, was durch andere Autoren bestätigt wurde.[46][47][48] Die Therapie zeigte bei einer Dosis von 12 Gray in Einzelfällen Erfolge.[49] In einer Langzeitstudie von 11 Jahren war bei einer durchschnittlichen Dosis von 38 Gray die Strahlentherapie nur bei einem Fall erfolgreich.[50] Bei einer neueren Untersuchung von 2004 zeigte sich eine Visusverbesserung von 0,28 auf 0,44, bei einer durchschnittlichen Tumorreduktion um ca. 40 %, wobei sich nicht alle Tumoren gleich stark verkleinerten. Die größte Reduktion zeigte sich bei kleineren Angiomen. Die Patienten wurden mit einer Gesamtdosis von 21,6 Gray fraktioniert über zwölf Tage bestrahlt. Als Nebenwirkung trat bei einem Patienten Katarakt auf.[51]
Protonentherapie
Bei dieser Therapiemethode besteht der Vorteil einer hohen Präzision der Gewebezerstörung, wobei nicht erkrankte Gewebeanteile geschont werden. Daher wird diese Methode angewendet, wenn Angiome in der Nähe von empfindlichen Gewebebereichen wachsen. Hierbei werden Protonen angewendet, die mittels großer Energie mehrere Zentimeter in das Gewebe eindringen können.[52][53]
Medikamentöse Behandlung
In klinischen Studien wurde die Behandlung der Angiome mit VEGF-Inhibitoren wie Pegaptanib[54] oder SU 5416[55][56] untersucht. Dabei wurden keine Veränderungen der Tumorgröße festgestellt. Jedoch traten Visusverbesserungen durch die Reduktion des Makulaödems auf. Als ernsthafte Nebenwirkung kann eine Anaphylaxie auftreten.[56] Zurzeit wird die medikamentöse Therapie als Begleittherapie neben den konventionellen Therapiemethoden diskutiert.[57][58]
Enukleation
Die Indikation zur Enukleation wird selten gestellt. Meist bei Schmerzen beim blinden Auge, bedingt durch ein Sekundärglaukom.[59]
Sonstige
Therapie ist die chirurgische Entfernung der Tumore.
Einzelnachweise
- J. van der Hoeve: The Doyne Memorial Lecturs: Eye symptoms in phakomatoses. In: Tr Ophth Soc UK. 1932. 52, S. 380.
- E. T. Collins: Two cases, brother and sister, with peculiar vascular new growth probably primarily retinal, affecting both eyes. In: Trans Opthalmol Soc. UK 1894, 14, S. 141–149.
- H. U. Moller: Familial angiomatosis retinae et cerebelli. In: Acta Ophthalmol. 1929, 7, S. 244–260.
- P. L. Choyke, G. M. Glenn, M. M. Walther u. a.: von Hippel–Lindau disease: genetic, clinical, and imaging features. In: Radiology. 1995, 194, S. 629–642.
- E. R. Maher, L. Iselius, J. R. Yates u. a.: von Hippel–Lindau disease: a genetic study. In: J Med Genet. 1991 28, S. 443–447.
- H. P. Neumann, O. D. Wiestler: Clustering of features of von Hippel–Lindau syndrome: evidence for a complex genetic locus. In: The Lancet. 1991, 337, S. 1052–1054.
- M. Ridley, J. Green and G. Johnson: Retinal angiomatosis: the ocular manifestations of von Hippel-Lindau disease. In: Can J Ophthalmol. 1986. 21(7), S. 276–283.
- J. T. Whitson, R. B. Welch, W. R. Green: Von Hippel-Lindau disease: case report of a patient with spontaneous regression of a retinal angioma. In: Retina. 1986. 6(4), S. 253–259.
- D. Schmidt, H. P. H. Neumann: Atypical retinal changes in v. Hippel-Lindau syndrome. In: Fortschr Ophthalmol. 1987, 84(2), S. 187–189.
- M. Bonnet, G. Garmier, S. Tlouzeau, C. Burtin: [Treatment of retinal capillary angiomas of von Hippels disease]. In: J Fr Ophtalmol. 1984, 7, S. 545–555.
- M. F. Goldberg, S. Koenig: Argon laser treatment of von Hippel–Lindau retinal angiomas. I. Clinical and angiographic findings. In: Arch Ophthalmol. 1974 92, S. 121–125.
- I. Kremer, E. Gilad, I. Ben-Sira: Juxtapapillary exophytic retinal capillary hemangioma treated by yellow krypton (568 nm) laser photocoagulation. In: Ophthalmic Surg. 1988, 19, S. 743–747.
- G. Meyer-Schwickerath: Lichtkoagulation bei Angiomatosis retinae. In: Lichtkoagulation, Bücherei des Augenarztes. 1959; 33, S. 62–70.
- J. D. M. Gass: Treatment of retinal vascular anomalies. In: Trans Am Acad Ophthalmol Otolaryngol. 1977, 83, S. 432–442.
- C. M. Lane, G. Turner, Z. J. Gregor, A. C. Bird: Laser treatment of retinal angiomatosis. In: Eye. 1989 3, S. 33–38.
- R. H. Rosa Jr, M. F. Goldberg, W. R. Green: Clinicopathologic correlation of argon laser photocoagulation of retinal angiomas in a patient with von Hippel–Lindau disease followed for more than 20 years. In: Retina. 1996, 16, S. 145–156.
- D. Schmidt, E. Natt, H. P. H. Neumann: Long-term results of laser treatment for retinal angiomatosis in von Hippel-Lindau disease. In: Eur J Med Res. 2000; 5, S. 47–58.
- C. F. Blodi, S. R. Russell, J. S. Pulido, J. C. Folk: Direct and feeder vessel photocoagulation of retinal angiomas with dye yellow laser. In: Ophthalmology. 1990, 97, S. 791–795; discussion 796–797.
- W. J. Annesley, B. C. Leonard, J. A. Shields, W. S. Tasman: Fifteen year review of treated cases of retinal angiomatosis. In: Trans Am Acad Ophthalmol Otolaryngol. 1977, 83, S. 446–453.
- A. Lommatzsch, A. Wessing: Angiomatosis retinae. Langzeitbeobachtungen. In: Ophthalmologe. 1996; 93, S. 158–162.
- G. A. Peyman, M. Raichand, R. C. Zeimer: Ocular effects of various laser wavelengths. In: Surv Ophthalmol. 1984, 28, S. 391–404.
- R. B. Welch: von Hippel–Lindau disease: the recognition and treatment of early angiomatosis retinae and the use of cryosurgery as an adjunct to therapy. In: Trans Am Ophthalmol Soc. 1970, 68, S. 367–424.
- A. Wessing: 10 Jahre Lichtkoagulation bei angiomatosis retinae. In: Klin Monatsbl Augenheilk. 1967. 150, S. 57–71.
- D. J. Apple, M. F. Goldberg, G. J. Wyhinny: Argon laser treatment of von Hippel-Lindau retinal angiomas. II. Histopathology of treated lesions. In: Arch Ophthalmol. 1974. 92(2), S. 126–130.
- I. Baras, S. Harris, M. A. Galin: Photocoagulation treatment of angiomatosis retinae. In: Am J Ophthalmol. 1964; 58, S. 296–299.
- M. F. Goldberg, J. R. Duke: Von Hippel-Lindau disease. Histopathologic findings in a treated and an untreated eye. In: Am J Ophthalmol. 1968; 66, S. 693–705.
- M. B. Gorin: von Hippel–Lindau disease: clinical considerations and the use of fluorescein-potentiated argon laser therapy for treatment of retinal angiomas. In: Semin Ophthalmol. 1992, 7, S. 182–191.
- H. Lincoff, J. McLean, R. Long: The cryosurgical treatment of intraocular tumors. Am J Ophthalmol 1967; 63, S. 389–399.
- R. C. Watzke: Cryotherapy for retinal angiomatosis. A clinicopathologic report. In: Arch Ophthalmol. 1974; 92, S. 399–401.
- J. A. Shields: Response of retinal capillary hemangioma to cryotherapy. In: Arch Ophthalmol. 1993; 111, S. 551.
- N. Bornfeld, K. M. Kreusel. Kapilläre Hämangiome der Netzhaut beim Von-Hippel-Lindau-Syndrom. In: Ophthalmologe. 2007; 104, S. 114–118.
- K. M. Kreusel, N. Bornfeld, A. Lommatzsch u. a. Ruthenium-106 brachytherapy for peripheral retinal capillary hemangioma. In: Ophthalmology. 1998; 105, S. 1386–1392.
- K. Rohrschneider, R. O. W. Burk, N. Bornfeld u. a. Kapilläres Hämangiom der Netzhaut. Laser-Scanning-tomographische Verlaufsbeobachtungen nach Strahlentherapie. In: Fortschr Ophthalmol. 1991; 88, S. 623–628.
- J. A. Oosterhuis, H. G. Journee-de Korver, H. M. Kakebeeke-Kemme, J. C. Bleeker: Transpupillary thermotherapy in choroidal melanomas. In: Arch Ophthalmol. 1995; 113, S. 315–321.
- C. L. Shields, M. C. Santos, W. Diniz u. a.: Thermotherapy for retinoblastoma. In: Arch Ophthalmol. 1999; 117, S. 885–893.
- I. S. Othmane, C. L. Shields, J. A. Shields u. a.: Circumscribed choroidal hemangioma managed by transpupillary thermotherapy. In: Arch Ophthalmol. 1999, 117, S. 136–137.
- C. L. Shields, J. A. Shields, J. Cater u. a.: Transpupillary thermotherapy for choroidal melanoma: tumor control and visual results in 100 consecutive cases. In: Ophthalmology. 1998, 105, S. 581–590.
- J. G. Journeé-De-Korver, J. A. Oosterhuis, H. M. Kakebeeke-Kemme u. a.: Thranspupillary thermotherapy (TTT) by infrared irradiation of choroidal melanoma. In: Doc Ophthalmol. 1992; 82, S. 185–191.
- D. N. Parmar, K. Mireskandari, D. McHugh: Transpupillary thermotherapy for retinal capillary hemangioma in von Hippel-Lindau disease. In: Ophthalm Surg Lasers. 2000; 31, S. 334–336.
- J. Garcia-Arum, L. H. Sararols, L. Cavero u. a.: Therapeutic options for capillary papillary hemangiomas. In: Ophthalmology. 2000; 107, S. 48–54.
- H. Rodriguez-Coleman, R. F. Spaide, L. A. Yannuzzi: Treatment of angiomatous lesions of the retina with photodynamic therapy. In: Retina. 2002; 22, S. 228–232.
- T. M. Aaberg, T. M. Aaberg, D. F. Martin u. a.: Three cases of large retinal capillary henangiomas treated with verteporfin and photodynamic therapy. In: Arch Ophthalmol. 2005; 123, S. 328–332.
- A. Szabo, Z. Géhl, A. Seres: Photodynamic (verteporfin) therapy for retinal capillary haemangioma, with monitoring of feeder and draining blood vessel diameters. In: Acta Ophthalmol Scand. 2005; 83, S. 512–513.
- S. J. Bakri, J. E. Sears, A. D. Singh. Transient closure of a retinal capillary hemangioma with verteporfin photodynamic therapy. In: Retina. 2005; 25, S. 1103–1104.
- A. W. M. Houwer: von Hippels disease: retinal angiomatosis. In: Am J Ophthalmol 1919; 2, S. 820.
- A. J. Ballantyne: Angiomatosis retinae. Account of a case including histologic results. In: Proc R Soc Med 1941; 35, S. 345–358.
- C. Cordes, M. J. Hogan: Angiomatosis retinae (Hippels disease): report of a case in which roentgen therapy was used in an early stage. In: Arch Ophthalmol 1940; 23, S. 253–269.
- F. H. McGovern: Angiomatosis retinae. In: Am J Ophthalmol 1943; 26, S. 184–187.
- F. C. Cordes, O. C. Dickson: Angiomatosis retinae (von Hippels disease). Results following irradiation of three eyes. In: Am J Ophthalmol. 1943; 26, S. 454–463.
- F. C. Cordes, A. Schwartz: Angiomatosis retinae (von Hippels disease) eleven years after irradiation. In: Trans Am Ophthalmol Soc. 1943; 50, S. 227–235.
- D. Raja, M. S. Benz,Murray TG u. a. Salvage external beam radiotherapy of retinal capillary hemangiomas secondary to von Hippel-Lindau disease. In: Ophthalmology. 2004; 111, S. 150–153.
- J. D. Palmer, E. S. Gragoudas: Advances in treatment of retinal angiomas. In: Int Ophthalmol Clin. 1997; 37, S. 150–170.
- N. Bornfeld, P. Chauvel, M. H. Foerster u. a. Protonentherapie intraokularer Tumoren. In: Klin Mbl Augenheilk. 1994; 204, S. 195.
- S. S. Dahr, M. Cusick, H. Rodriguez-Coleman u. a. Intravitreal anti-vascular endothelial growth factor therapy with Pegaptanib for advanced von Hippel-Lindau disease of the retina. In: Retina. 2007; 27, S. 150–158.
- J. F. Girmens, A. Erginay, P. Massin u. a. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. In: Am J Ophthalmol. 2003; 136, S. 194–196.
- L. P. Aiello, D. J. George, M. T. Cahill u. a. Rapid and durable recovery of visual function in a patient with von Hippel-Lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor SU5416. In: Ophthalmology. 2002; 109, S. 1745–1751.
- S. Madhusudan, G. Deplanque, J. P. Braybrooke u. a. Antiangiogenic therapy for von Hippel-Lindau disease. In: JAMA. 2004; 291, S. 943–944.
- M. I. Rosenblatt, D. T. Azar. Anti-angiogenic therapy: prospects for treatment of ocular tumors. In: Seminars Ophthalmol. 2006; 21, S. 151–160.
- A. R. Webster, E. R. Maher, A. T. Moore: Clinical characteristics of ocular angiomatosis in von Hippel–Lindau disease and correlation with germline mutation. In: Arch Ophthalmol. 1999; 117, S. 371–378.
Literatur
Bücher
- Olaf Rieß, Ludger Schöls (Hrsg.): Neurogenetik. Molekulargenetische Diagnostik neurologischer Erkrankungen. Springer. Berlin 1998, ISBN 3-540-63874-1.
- P. Lewis. Rowland (Hrsg.): Merrits Textbook of Neurology. Williams and Wilkins. Baltimore 1995, ISBN 0-683-07400-8.
- S. Mark. Greenberg: Handbook of Neurosurgery. Lakeland 1997, ISBN 0-9626384-5-5.
- O. Bruce. Berg (Hrsg.): Principles of Child Neurology. McGraw-Hill. New York 1996, ISBN 0-07-005193-3.
- D. Raymond. Adams (Hrsg.): Principles of Neurology. McGraw-Hill. New York 1997, ISBN 0-07-067439-6.
Fachartikel
- J. P. Constans u. a.: Posterior fossa hemangioblastomas. In: Surgical Neurology. 1986, 25, S. 269–275. PMID 3945908
- D. M. Hough u. a.: Pancreatic lesions in von Hippel-Lindau diseas: Prevalence, clinical significance and CT findings. In: Am J Radiol. 1994, 162, S. 1091–1094. PMID 8165988
- E. R. Maher u. a.: Familial renal cell carcinoma. Clinical and molecular genetic aspects. In: Br J Cancer. 1991; 63, S. 176–179. PMID 1997093
- E. R. Maher u. a.: Von Hippel-Lindau disease: a genetic study. In: J Med Genet. 1991, 28, S. 443–447. PMID 1895313
- K. L. Melmon u. a.: Lindaus disease: Review of the literature and study of a large kindred. In: Am J Med. 1964, 36, S. 595–617. PMID 14142412
- H. P. H. Neumann u. a.: Clustering of features of Hippel-Lindau syndrom. In: The Lancet. 1991; 337, S. 1062–1054. PMID 1673491
- H. P. H. Neumann u. a.: Von Hippel Lindau Syndrom. In: Brain Pathol. 1995; 5, S. 181–193. PMID 7670659
- J. M. Whaley u. a.: Germ-line mutations in the von Hippel-Lindau tumor-suppressor gene are similar to somatic von Hippel-Lindau aberrations in sporadic renal cell carcinoma. In: Am J Hum Genet. 1994 Dec;55(6), S. 1092–1102. PMID 7977367