Interleukin-17
Interleukin-17-Familie (IL17-Familie) Die Interleukin-17-Zytokine bilden eine Familie proinflammatorischer Zytokine mit einer besonderen Struktur. IL-17-Familienmitglieder besitzen vier Cystein-Glieder, die zwei Disulfid-Brücken bilden (Cystein-Knoten).[2] Sie werden von Th17-Helferzellen nach einer Stimulation mit Interleukin-23 gebildet und sezerniert.[3] Biologisch aktive IL-17-Zytokine reagieren mit spezifischen Oberflächenrezeptoren (IL17RA, IL17RB, IL17RC) der Zielzellen. Die Rezeptorbindung kann in den Zielzellen eine Kaskade weiterer Zytokine und Chemokine auslösen, die insbesondere Immunzellen, wie Monozyten und neutrophile Granulozyten, anlocken.[4] Die IL-17-Familie ist an der Pathogenese vieler entzündlicher und autoimmuner Erkrankungen beteiligt. Zu nennen sind rheumatoide Arthritis[5], Asthma[6], Psoriasis[7][8], Lupus erythematodes und Colitis ulcerosa[9]. Auch an der Allograft-Abstoßung und der antitumoralen Immunität sind IL-17-Zytokine beteiligt.[7]
| Interleukin-17A | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 35kDa | |
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17A IL17, CTLA8 | |
| Externe IDs | ||
| Vorkommen | ||
| Interleukin-17B | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17B ZCOTO7 | |
| Externe IDs | ||
| Vorkommen | ||
| Interleukin-17C | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17C CX2 | |
| Externe IDs | ||
| Vorkommen | ||
| Interleukin-17D | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17D IL17, CTLA8 | |
| Externe IDs | ||
| Vorkommen | ||
| Interleukin-17E | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17E Interleukin-25, IL25 | |
| Externe IDs | ||
| Vorkommen | ||
| Interleukin-17F | ||
|---|---|---|
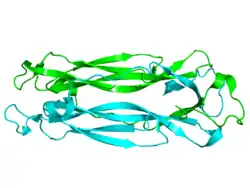
| ||
| Kristallographische Struktur des dimeren humanen IL-17f.[1] | ||
| Eigenschaften des menschlichen Proteins | ||
| Sekundär- bis Quartärstruktur | Dimer | |
| Bezeichner | ||
| Gen-Namen | IL17F ML-1 | |
| Externe IDs | ||
| Vorkommen | ||
Entdeckungsgeschichte
Im Jahr 1993 isolierten Rouvier und Mitarbeiter das IL17A-Gen aus T-Zell-Hybriden von Nagern.[10] Das codierte Protein war das Gründungsmitglied IL-17A der IL-17-Familie. Das Nager-Protein wies eine starke Homologie mit einem Virus-Genprodukt auf. Es handelt sich um das Herpesvirus saimiri, ein T-Zell-zytotrophes Virus, welches auch unter der Bezeichnung CTLA8 beschrieben wird.[11]
Struktur
IL-17-A ist ein Protein aus 155 Aminosäuren. Über zwei Disulfid-Brücken bildet es Homodimere oder mit IL-17F Heterodimere. Die dazu erforderlichen vier Cysteine sind hoch konserviert. Es wird als Glykoprotein sezerniert und hat eine Masse von 35 kDa.[12] Die Untereinheiten der Dimere haben eine Masse von 12–20 kDa. Ein Bereich von 23 Aminosäuren fungiert als Signalstruktur. Es folgt eine 123 Aminosäuren langer Abschnitt, der charakteristisch für die IL-17-Familie ist. Die Glykolisierung besteht bei 15 und 20 kDa über eine N-Bindung.[13] IL-17 hat keinerlei Ähnlichkeit mit anderen bekannten Interleukine. Auch gibt es keinerlei Entsprechungen zu anderen bekannten Proteinen oder strukturellen Domänen.[14]
Die Kristallstruktur von IL-17F, welches zu 50 % homolog zu IL-17A ist, zeigt eine Ähnlichkeit mit der Cystein-Knoten-Familie, zu der auch Neurotrophine des ZNS gehören. Die Cystein-Knoten-Faltung ist charakterisiert durch zwei paarige β-Ketten. die durch drei Disulfidbrücken stabilisiert werden. Im Gegensatz zu den anderen Cytein-Knoten-Proteinen fehlt IL-17F die dritte Disulfidbrücke. An der entsprechenden Stelle ist Cystein durch Serin ersetzt. Diese Besonderheit ist bei allen Mitgliedern der IL-17-Familie konserviert. IL-17F dimerisiert ähnlich wie der Nerve growth factor (NGF) und andere Neurotropine.[1]
Mitglieder der IL-17-Familie
Die IL-17-Familie besteht beim Menschen aus:
- IL-17A: z. T. fälschlich als IL-17 bezeichnet
- IL-17B
- IL-17C
- IL-17D
- IL-17E: auch als Interleukin-25 bezeichnet
- IL-17F: Homologie mit IL-17A
Alle haben eine ähnliche Proteinstruktur. Die Aminosäuresequenz enthält vier hoch konservierte Cystein-Baustein, die entscheidend für die richtige 3-dimensionale Gestalt des ganzen Proteins ist. Bei der Sequenz gibt es keine signifikante Homologie zu anderen Zytokinen.
Innerhalb der Familie haben die IL-17F-Isoformen 1 und 2 (ML-1) die höchste Sequenz-Homologie mit IL-17A (55 % und 40 %). Beide Zytokine werden von denselben Zelltypen exprimiert, zum Beispiel Th17-Helferzellen, γδ-T-Zellen und unreife lymphoide Zellen. Sie können als Homodimere oder Heterodimere IL-17A - IL-17F sezerniert werden und aktivieren die Signalkaskade durch denselben Membranrezeptor-Komplex IL-17RA/IL-17RC.[15]
IL-17B hat 29 % Homologie mit IL-17-A, 25 % mit IL-17-D und 23 % mit IL-17-C und 17 % mit IL-17-E. Innerhalb der Säugetiere ist die Sequenz stark konserviert. Die Homologie der IL-17-Moleküle zwischen humanen und analogen Proteinen der Maus beträgt 62–88 %.[12]
Gene
Das Gen, welches IL-17A kodiert, ist 1874 Basenpaare lang und wurde aus CD4+ T-Zellen geklont.[13] Die Gen-Expression der verschiedenen IL-17-Zytokine ist unterschiedlich. Die Gene von IL-17A und IL-17F scheinen nur von einer kleinen Gruppe aktivierter T-Zellen exprimiert und bei Entzündungen hochreguliert zu werden. Das Gen von IL-17B wird von mehreren peripheren Geweben und von Immungeweben exprimiert. IL-17C wird ebenfalls bei Entzündungen hochreguliert. Im Ruhezustand ist die Genaktivität minimal oder fehlend. Das Gen von IL-17D wird stark im Nervensystem und in Skelettmuskeln exprimiert. Das IL-17E-Gen wird in verschiedenen peripheren Geweben in geringer Konzentration gefunden. Das humane IL-17F-Gen wurde 2001 entdeckt und ist auf dem Chromosom 6p12 lokalisiert.
IL-17-Rezeptoren
Die Il-17-Rezeptorfamilie besteht aus 5 weit verbreiteten Rezeptoren, die sich in ihrer Ligandenspezifität unterscheiden:
IL-17RA wird in verschiedenen Geweben exprimiert, zum Beispiel Gefäßendothel, periphere T-Zellen, verschiedene B-Zelllinien, Fibroblasten, Lungengewebe, myelomonozytäre Zellen, Knochenmarksstromazellen.[12][16][2] Für die Auslösung des Il-17A- oder Il-17F-Signals ist ein heterodimer Komplex aus IL-17RA und IL-17RC erforderlich. Fehlt eine Komponente, so kommt es nicht zur Signaltransduktion. Analog benötigt IL-17E einen heterodimeren Komplex aus IL-17RA und IL-17RB. Der Komplex IL-17RA-IL-17RB wird auch als IL-17Rh1, IL-17BR oder IL-25R beschrieben.[17]
IL-17RB wird in der Niere, dem Pankreas, der Leber, dem Gehirn und dem Darm exprimiert.[12]
IL-17RC wird in der Prostata, dem Knorpel, der Niere, der Leber, dem Herzen und in der Muskulatur exprimiert. Durch alternatives Spleißen kann ein lösliche Rezeptor zusätzlich zu der Zellmembran-gebundenen Form erzeugt werden.
Auch das Gen für IL-17RD kann durch alternatives Spleißen einen löslichen Rezeptor bilden. Lösliche Rezeptoren binden IL-17, ohne ein Signal auszulösen, vermindern also die proinflammatorische Wirkung von IL-17.[12][2]
IL-17RE wird im Pankreas, dem Gehirn und der Prostata exprimiert.[12]
Die Signaltransduktion dieser Rezeptoren ist nur teilweise erforscht. Innerhalb der Rezeptoren ist sie so verschieden wie deren Verteilung im Körper. Auch weisen die verschiedenen Rezeptoren keine signifikante Ähnlichkeit im extrazellulären oder intrazellulären Anteil im Vergleich mit anderen Zytokin-Rezeptoren aus.[16] An der Signaltransduktion sind insbesondere Transkriptionen Faktoren wie TRAF6, JNK, Erk1/2, p38, AP-1 and NF-κB beteiligt. Die Wirkung ist abhängig vom Gewebe und dem Aktivierungszustand der Zellen.[16][2][18]
Funktion und Regulation
Zahlreiche immunregulatorische Funktionen der IL-17-Familie wurden publiziert. Die Effekte beruhen auf der Induktion von immunologischen Signalmolekülen. Die wichtigste Rolle spielen IL-17-Zytokine bei der Auslösung und Vermittlung eines proinflammatorischen Respons, also der Auslösung und Steigerung einer Entzündung (mit oder ohne Erreger). Sie sind auch an allergischen Reaktionen beteiligt. Typischer Auslöser der Signalkaskade ist die Invasion von Krankheitserregern in den Körper. Bei der entzündlichen Abwehrreaktion kooperiert IL-17 häufig mit dem Tumornekrosefaktor und Interleukin-1.[19][20]
IL-17-Zytokine induzieren folgende Substanzen:
- IL-6
- NF-κB[21]
- G-CSF
- GM-CSF
- IL-1β
- TGF-β
- TNF-α
- Chemokine inklusive IL-8
- GRO-α
- MCP-1
- Prostaglandine zum Beispiel PGE2
Die Induktion kann durch folgende Zelltypen erfolgen:
- Fibroblasten
- Endothelzellen
- Epithelzellen zum Beispiel Keratinozyten
- Makrophage
Gemeinsam mit Interleukin-22 induzieren IL-17-Zytokine bei Keratinozyten die Synthese antimikrobiellen Proteine. Interleukin-22 wird beim Menschen durch T22-Helferzellen, bei Mäusen durch T17-Helferzellen produziert. In der Atemwegen sind IL-17-Zytokine am Remodeling beteiligt. Durch die gesteigerte Chemokinexpression werden Zellen angelockt, insbesondere neutrophile Granulozyten, nicht aber eosinophile Granulozyten. IL-17 ist essentiell für T17-Helferzellen.
Die aktive Form von Vitamin D soll die Produktion von Il-17-Zytokinen und deren Rezeptoren durch T-Helferzellen vermindern.[22]
IL-17 und andere Zytokine
Als Teil einer komplexen proentzündlichen Signalkaskade ist IL-17 von zahlreichen anderen Signalmolekülen abhängig oder kooperiert mit ihnen. Aggarwal und Mitarbeiter konnten zeigen, dass die Produktion von IL-17 von Interleukin-23 abhängig ist.[3] Später entdeckte eine koreanische Arbeitsgruppe, dass der STAT3- und NF-κB-Signalweg für diese IL-23-vermittelte IL-17-Produktion erforderlich ist.[23] Chen und Mitarbeiter wiesen nach, dass noch ein weiteres Molekül, SOCS3, eine bedeutende Rolle bei der IL-17-Produktion spielt.[24] Fehlt SOCS3, so wird die IL-23-induzierte STAT3-Phosphorylierung hochgefahren, und das phosphorylierte STAT3 bindet an die Promotorregion von IL-17A und IL-17F und erhöht so deren Genaktivität. Es gibt allerdings auch Wissenschaftler, die glauben, dass die IL-17-Induktion unabhängig von IL-23 möglich ist. Bei in vitro-Experimenten verwendeten die Arbeitsgruppen TGF-β und IL-6[25] and in vivo[26][27][25][26][27] Auch wenn IL-23 für die Expression von IL-17 nicht essenziell ist, so mag es doch eine Rolle beim Überleben und der Proliferation der IL-17-produzierenden T-Zellen spielen. Die Kooperation von IL-17A und IL-17F mit dem Tumornekrosefaktor (TNF) konnte an Zellkulturen von Fibroblasten und Synoviozyten von Patienten mit Psoriasis-Arthritis gezeigt werden. Die gleichzeitige Gabe von TNF und IL-17A oder IL-17F induzierte ein Vielfaches an Interleukin 8 wie die Gabe von IL-17A oder IL-17F allein.[28]
Die Differenzierung von IL-17 produzierenden T-Zellen im Thymus wird von RAR-related orphan receptor gamma (ROR-γ), einem spezifischen Kernrezeptor, gesteuert.[29]
IL-17 in der Pathogenese von Erkrankungen
Durch zahlreiche Funktionen sind IL-17-Zytokine an der Pathogenese immunologischer und autoimmunologischer Krankheiten beteiligt, zum Beispiel rheumatoide Arthritis, Asthma, Lupus erythematodes, Allograft-Abstoßung, Antitumorale Immunität und Psoriasis,[14] Multiple Sklerose und intracerebrale Hämorrhagie.[30]
IL-17 und Psoriasis
Der IL-23 / IL-17-Signalweg spielt eine wichtige Rolle in der Pathogenese der Schuppenflechte.[7][8][31] Bei dieser Erkrankung reagieren Immunzellen auf proinflammatorische Moleküle, die in der Haut, dem Skalp und der Umgebung der Gelenke freigesetzt werden.[8] Die Folge ist eine Beschleunigung der Zellteilung epidermaler Hautzellen, was zur Bildung von roten, schuppigen Läsionen und einer chronischer Entzündung der Haut führt.[31][32] An Hautbiopsien von Psoriasis-Läsionen konnte eine Anreicherung toxischer T-Zellen und neutrophiler Granulozyten nachgewiesen werden, die IL-17 enthielten.[8][33][34] Das ist ein Hinweis darauf, dass eine exzessive Infiltration proinflammatorische Immunzellen und Il-17-Zytokine mit der Entwicklung der Psoriasis zusammenhängen.
Bei Tierversuchen mit Mäusen konnte gezeigt werden, dass die Blockierung von IL-23 oder IL-17 das Fortschreiten der Psoriasis verminderte.[35][36]
Injizierte man Mäusen monoclonale Antikörper gegen IL-17, so wurde dieses Zytokin blockiert oder neutralisiert. Die Down-Stream-Signale wurden vermindert und die epidermale Hyperplasie ging zurück.[35] Bei genetisch modifizierten Mäusen, die keine Rezeptoren für IL-23 oder IL-17 exprimierten, verursachte der auslösende Promotor, Phorbol-12-myristat-13-acetat, eine geringere psoriasisartige Läsion.[8][36] Die Entzündung beginnt, wenn Keratinozyten das letzte Stadium ihres Zellzyklus erreicht haben, was unreife dendritischer Zellen (DC) aktiviert.[37] Die DCs sezernieren Zytokine, welche die degenerierenden Keratinozyten veranlasst, TNF-alpha, Interleukin-1 und Interleukin-6 zu produzieren. Dadurch wird ein chemotaktisches Signal erzeugt, welches T-Zellen, natürliche Killerzellen und Monozellen in die Epidermis anlockt.[32] Diese angelockten Zellen bilden IL-23, welches bei Th17-Helferzellen die Produktion von IL-17 induziert.[33]
IL-17 reagiert mit IL-17RA-Rezeptoren, welche reichlich auf der Oberfläche der Keratinozyten vorkommen. Diese bilden daraufhin vermehrt IL-6, antimikrobielle Peptide, IL-8 und CCL20.[7][31][36] Die erhöhte Konzentration von Il-6 vermindert die Fähigkeit von regulatorischen T-Zellen (T-reg), das Verhalten von Th-17-Helferzellen zu kontrollieren.[33] Die verminderte Regulation erlaubt den Th17-Helferzellen, sich ungehindert zu vermehren, und in den Psoriasisherden noch mehr IL-17 zu produzieren. Insgesamt wird das IL-17 Signal sehr verstärkt.[33] Antimikrobielle Peptide und IL-8 locken neutrophile Granulozyten in die Läsion. Dort eliminieren die Granulozyten defekte und entzündlich gereizte Keratinozyten.[8][34][36] Durch CCL20 werden weitere unreife dendritische Zellen via Chemotaxis in die Psoriasisherde gelockt, wo ihre Aktivierung erfolgt und die proinflammatorische Kaskade verstärkt wird.[33][34] IL-17 und andere Zytokine, die von den eingewanderten neutrophilen Granulozyten, T-Zellen und dendritischen Zellen sezerniert werden, wirken auf die lokalen Leukozyten und Keratinozyten ein, so dass die Psoriasis fortschreitet und eine chronische Entzündung unterhalten wird.[33]
Die Rolle von IL-17 bei Asthma
Aus der IL-17-Familie wurde für IL-17F in vitro und in vivo gezeigt, dass es eine proinflammatorischer Rolle bei Asthma spielt. IL-17F wird in den Atemwegen von Asthmatikern exprimiert und die Konzentration korreliert mit dem Schweregrad der Erkrankung. Darüber hinaus ist die Code-Variante H161R des IL-17F-Gens invers mit Asthma korreliert. Sie codiert einen Antagonisten für den Wildtyp von IL-17F. In den Atemwegen kann IL-17F zahlreiche Zytokine, Zytokine und Adhäsionsmoleküle in Zellen des Bronchialepithels, des venösen Endothels, in Fibroblasten und eosinophilen Granulozyten induzieren. Dabei bindet IL-17F an die Rezeptoren IL-17RA und IL-17RC und aktiviert den MAPK/ERG-Signalweg. Das IL-17F der Atemwege stammt aus Th17-Helferzellen, Mastzellen, basophilen Granulozyten und wird auch in den Geweben, inklusive der Lunge, exprimiert. Eine Überexpression des IL-17F-Gens der Atemwege von Mäusen ist mit einer Infiltration der Luftwege mit neutrophilen Granulozyten, der Induktion vieler Zytokine, einer Hyperreaktivität der Bronchien und einer vermehrten Schleimsekretion verbunden.
Zusammenfassend kann man sagen, dass IL-17F eine zentrale Rolle bei allergischen Atemwegserkrankungen spielt und eine große Bedeutung bei der Behandlung von Asthma haben könnte.[6]
Sonstige Erkrankungen
IL-17 spielt möglicherweise eine Rolle bei der Progression der multiplen Sklerose.[38]
Auf der Basis von Tierversuchen wurde vorgeschlagen, Il-17-Inhibitoren nach Schlaganfällen einzusetzen, um sekundäre entzündliche Prozesse zu bremsen.[39]
Chronische Entzündungen können das Risiko von malignen Erkrankungen erhöhen. Der Mechanismus ist weitgehend unbekannt. Ortitz und Mitarbeiter vermuten, dass unreife myeloide Zellen an der Entstehung von Hautkrebs beteiligt sind. Diese rekrutieren CD4+ T-Zellen, die IL-17 produzieren.[40]
Pharmakologische IL-17-Inhibitoren
Wegen der Beteiligung an der Immunregulation, wurden IL-17Inhibitoren auf ihre Wirksamkeit bei Autoimmunerkrankungen wie rheumatoide Arthritis, Psoriasis und entzündliche Darmerkrankungen untersucht.[41][42][43]
Secukinumab
Monoclonalen Antikörper, der Il-17A inhibiert. In der randomisierten Studie "FUTURE 1" konnte gezeigt werden, dass Secukinumab die Symptome einer Psoriasis verbessert. Im Januar 2015 erfolgte die FDA-Zulassung von Secukinumab für die Behandlung von moderater oder schwerer Plaque-Psoriasis. Es wurde von der Firma Novartis entwickelt und unter dem Namen Cosentyx® vertrieben. Es wurde in Japan für die Behandlung der Psoriasis-Arthritis zugelassen.[44] Die europäische EMA hat Secukinumab für folgende Indikationen zugelassen:
- Psoriasis im Erwachsenenalter
- Psoriasis bei Kindern ab sechs Jahre
- aktive, psoriatrische Arthritis: allein oder in Kombination mit Methotrexat, wenn die Wirkung von DMARDs (disease-modifying anti-rheumatic drug) nicht ausreicht.
- Ankylosierende Spondylitis: bei ungenügender Wirkung konventioneller Therapien
- Axiale Spondylarthritis ohne Röntgenzeichen, (englisch: Non-radiographic axial spondyloarthritis, nr-axSpA)[45]
Die FDA hat Secukinumab analog zugelassen.[46]
Ixekizumab
Ebenfalls ein monoklonaler Antikörper gegen IL-17A, der von der Firma Elli Lily unter dem Handelsnamen Taltz® vertrieben wird. In drei randomisierten Studien (UNCOVER-1, UNCOVER-2,UNCOVER-3) konnte gezeigt werden, dass Ixekizumab die Symptome einer Psoriasis verbessert. An Nebenwirkungen wurden Neutropenien, Pilzinfektionen und entzündliche Darmerkrankungen beobachtet.[47] Die von der EMA genehmigten Indikationen entsprechen denen von Secukinumab.[48]
Bimekizumab
Dieser monoklonale Antikörper blockiert gleichzeitig IL-17A und IL-17F. An Zellkulturen von Fibroblasten und Synoviozyten von Patienten mit Psoriasis-Arthritis konnte gezeigt werden, dass die gleichzeitige Blockade von IL-17A und IL-17F die Produktion von Interleukin 8 stärker suprimiert als jedes allein.[28] Die EMA hat das Medikament unter dem Handelsnamen Bimekx® zur Behandlung der mittelschweren und schweren Plaque-Psoriasis zugelassen.[49] In der Studie "BE SURE" konnte gezeigt werden, dass die Wirkung bei Psoriasis mindestens gleich gut, wie die des TNF-Inhibitors Adalimumab ist. An Nebenwirkungen traten vermehrt Pilzinfektionen der Mundhöhle und Durchfälle auf.[50]
Brodalumab
Dieser von der Firma Amgen unter dem Namen Kyntheum® vertriebene monoklonale Antikörper richtet sich nicht gegen IL-17, sondern gegen den Rezeptor IL-17RA. In einer randomisierten Studie (ClinicalTrials.gov number: NCT01516957) konnte gezeigt werden, dass Brodalumab die Symptome einer Psoriasis verbessert.[51] Im Jahr 2017 erteilte die EMA die Zulassung für die Behandlung der Psoriasis.[52]
Ustekinumab
Monoklonaler Antikörper, der Interleukin - 23 blockiert. Das führt indirekt zu einer Verminderung von IL-17. Daher kann es ebenfalls effektiv bei der Behandlung der Psoriasis eingesetzt werden.[53]
Einzelnachweise
- Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, Pan G, Gurney AL, de Vos AM, Starovasnik MA: IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. In: The EMBO Journal. Band 20, Nr. 19, Oktober 2001, S. 5332–41, doi:10.1093/emboj/20.19.5332, PMID 11574464, PMC 125646 (freier Volltext).
- Moseley TA, Haudenschild DR, Rose L, Reddi AH: Interleukin-17 family and IL-17 receptors. In: Cytokine & Growth Factor Reviews. Band 14, Nr. 2, April 2003, S. 155–74, doi:10.1016/S1359-6101(03)00002-9, PMID 12651226.
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL: Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. In: The Journal of Biological Chemistry. Band 278, Nr. 3, Januar 2003, S. 1910–4, doi:10.1074/jbc.M207577200, PMID 12417590.
- Starnes T, Broxmeyer HE, Robertson MJ, Hromas R: Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. In: Journal of Immunology. Band 169, Nr. 2, Juli 2002, S. 642–6, doi:10.4049/jimmunol.169.2.642, PMID 12097364 (jimmunol.org).
- Myew-Ling Toh, Gaelle Gonzales, Marije I. Koenders, Anne Tournadre, David Boyle: Role of Interleukin 17 in Arthritis Chronicity through Survival of Synoviocytes via Regulation of Synoviolin Expression. In: PLoS ONE. Band 5, Nr. 10, 15. Oktober 2010, ISSN 1932-6203, S. e13416, doi:10.1371/journal.pone.0013416, PMID 20976214, PMC 2955522 (freier Volltext) – (plos.org [abgerufen am 13. September 2021]).
- Kawaguchi M, Kokubu F, Fujita J, Huang SK, Hizawa N: Role of interleukin-17F in asthma. In: Inflammation & Allergy Drug Targets. Band 8, Nr. 5, Dezember 2009, S. 383–9, doi:10.2174/1871528110908050383, PMID 20025586.
- Martin DA, Towne JE, Kricorian G, Klekotka P, Gudjonsson JE, Krueger JG, Russell CB: The emerging role of IL-17 in the pathogenesis of psoriasis: preclinical and clinical findings. In: The Journal of Investigative Dermatology. Band 133, Nr. 1, Januar 2013, S. 17–26, doi:10.1038/jid.2012.194, PMID 22673731, PMC 3568997 (freier Volltext).
- Lowes MA, Suárez-Fariñas M, Krueger JG: Immunology of psoriasis. In: Annual Review of Immunology. Band 32, 2014, S. 227–55, doi:10.1146/annurev-immunol-032713-120225, PMID 24655295, PMC 4229247 (freier Volltext).
- Stephan R Targan, Brian Feagan, Severine Vermeire, Remo Panaccione, Gil Y Melmed: A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. In: American Journal of Gastroenterology. Band 111, Nr. 11, November 2016, ISSN 0002-9270, S. 1599–1607, doi:10.1038/ajg.2016.298 (lww.com [abgerufen am 13. September 2021]).
- Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K: Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. In: The British Journal of Dermatology. Band 160, Nr. 2, Februar 2009, S. 319–24, doi:10.1111/j.1365-2133.2008.08902.x, PMID 19016708.
- Rouvier E, Luciani MF, Mattéi MG, Denizot F, Golstein P: CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. In: Journal of Immunology. Band 150, Nr. 12, Juni 1993, S. 5445–56, PMID 8390535 (jimmunol.org).
- Kolls JK, Lindén A: Interleukin-17 family members and inflammation. In: Immunity. Band 21, Nr. 4, Oktober 2004, S. 467–76, doi:10.1016/j.immuni.2004.08.018, PMID 15485625.
- Yao Z, Painter SL, Fanslow WC, Ulrich D, Macduff BM, Spriggs MK, Armitage RJ: Human IL-17: a novel cytokine derived from T cells. In: Journal of Immunology. Band 155, Nr. 12, Dezember 1995, S. 5483–6, PMID 7499828 (jimmunol.org).
- Aggarwal S, Gurney AL: IL-17: prototype member of an emerging cytokine family. In: Journal of Leukocyte Biology. Band 71, Nr. 1, Januar 2002, S. 1–8, PMID 11781375.
- Sophie Glatt, Dominique Baeten, Terry Baker, Meryn Griffiths, Lucian Ionescu: Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. In: Annals of the Rheumatic Diseases. Band 77, Nr. 4, April 2018, ISSN 0003-4967, S. 523–532, doi:10.1136/annrheumdis-2017-212127, PMID 29275332, PMC 5890624 (freier Volltext) – (bmj.com [abgerufen am 13. September 2021]).
- Kawaguchi M, Adachi M, Oda N, Kokubu F, Huang SK: IL-17 cytokine family. In: The Journal of Allergy and Clinical Immunology. Band 114, Nr. 6, Dezember 2004, S. 1265–73; quiz 1274, doi:10.1016/j.jaci.2004.10.019, PMID 15577820.
- Pappu R, Ramirez-Carrozzi V, Sambandam A: The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. In: Immunology. Band 134, Nr. 1, September 2011, S. 8–16, doi:10.1111/j.1365-2567.2011.03465.x, PMID 21726218, PMC 3173690 (freier Volltext).
- Ley K, Smith E, Stark MA: IL-17A-producing neutrophil-regulatory Tn lymphocytes. In: Immunologic Research. Band 34, Nr. 3, 2006, S. 229–42, doi:10.1385/IR:34:3:229, PMID 16891673.
- Chiricozzi A, Guttman-Yassky E, Suárez-Fariñas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG: Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. In: The journal of Investigative Dermatology. Band 131, Nr. 3, März 2011, S. 677–87, doi:10.1038/jid.2010.340, PMID 21085185.
- Miossec P, Korn T, Kuchroo VK: Interleukin-17 and type 17 helper T cells. In: The New England Journal of Medicine. Band 361, Nr. 9, August 2009, S. 888–98, doi:10.1056/NEJMra0707449, PMID 19710487.
- M. Awane, P. G. Andres, D. J. Li, H. C. Reinecker: NF-kappa B-inducing kinase is a common mediator of IL-17-, TNF-alpha-, and IL-1 beta-induced chemokine promoter activation in intestinal epithelial cells. In: Journal of Immunology (Baltimore, Md.: 1950). Band 162, Nr. 9, 1. Mai 1999, ISSN 0022-1767, S. 5337–5344, PMID 10228009 (nih.gov [abgerufen am 13. September 2021]).
- Chang SH, Chung Y, Dong C: Vitamin D suppresses Th17 cytokine production by inducing C/EBP homologous protein (CHOP) expression. In: The Journal of Biological Chemistry. Band 285, Nr. 50, Dezember 2010, S. 38751–5, doi:10.1074/jbc.C110.185777, PMID 20974859, PMC 2998156 (freier Volltext).
- Cho ML, Kang JW, Moon YM, Nam HJ, Jhun JY, Heo SB, Jin HT, Min SY, Ju JH, Park KS, Cho YG, Yoon CH, Park SH, Sung YC, Kim HY: STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. In: Journal of Immunology. Band 176, Nr. 9, Mai 2006, S. 5652–61, doi:10.4049/jimmunol.176.9.5652, PMID 16622035 (jimmunol.org).
- Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ: Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. In: Proceedings of the National Academy of Sciences of the United States of America. Band 103, Nr. 21, Mai 2006, S. 8137–42, doi:10.1073/pnas.0600666103, PMID 16698929, PMC 1459629 (freier Volltext), bibcode:2006PNAS..103.8137C.
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B: TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. In: Immunity. Band 24, Nr. 2, Februar 2006, S. 179–89, doi:10.1016/j.immuni.2006.01.001, PMID 16473830.
- Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT: Transforming growth factor-beta induces development of the T(H)17 lineage. In: Nature. Band 441, Nr. 7090, Mai 2006, S. 231–4, doi:10.1038/nature04754, PMID 16648837.
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK: Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. In: Nature. Band 441, Nr. 7090, Mai 2006, S. 235–8, doi:10.1038/nature04753, PMID 16648838.
- Sophie Glatt, Dominique Baeten, Terry Baker, Meryn Griffiths, Lucian Ionescu: Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. In: Annals of the Rheumatic Diseases. Band 77, Nr. 4, April 2018, ISSN 0003-4967, S. 523–532, doi:10.1136/annrheumdis-2017-212127, PMID 29275332, PMC 5890624 (freier Volltext) – (bmj.com [abgerufen am 13. September 2021]).
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR: The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. In: Cell. Band 126, Nr. 6, September 2006, S. 1121–33, doi:10.1016/j.cell.2006.07.035, PMID 16990136.
- Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, Che F, Chen X, Ren H, Hong M, Wang J: Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. In: Progress in Neurobiology. Band 178, Juli 2019, S. 101610, doi:10.1016/j.pneurobio.2019.03.003, PMID 30923023.
- Hu Y, Shen F, Crellin NK, Ouyang W: The IL-17 pathway as a major therapeutic target in autoimmune diseases. In: Annals of the New York Academy of Sciences. Band 1217, Nr. 1, Januar 2011, S. 60–76, doi:10.1111/j.1749-6632.2010.05825.x, PMID 21155836.
- Baliwag J, Barnes DH, Johnston A: Cytokines in psoriasis. In: Cytokine. Band 73, Nr. 2, Juni 2015, S. 342–50, doi:10.1016/j.cyto.2014.12.014, PMID 25585875, PMC 4437803 (freier Volltext).
- Mudigonda P, Mudigonda T, Feneran AN, Alamdari HS, Sandoval L, Feldman SR: Interleukin-23 and interleukin-17: importance in pathogenesis and therapy of psoriasis. In: Dermatology Online Journal. Band 18, Nr. 10, Oktober 2012, S. 1, doi:10.5070/D33N39N8XM, PMID 23122008 (escholarship.org).
- Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, Villanueva EC, Shah P, Kaplan MJ, Bruce AT: Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. In: Journal of Immunology. Band 187, Nr. 1, Juli 2011, S. 490–500, doi:10.4049/jimmunol.1100123, PMID 21606249, PMC 3119764 (freier Volltext).
- Nakajima K, Kanda T, Takaishi M, Shiga T, Miyoshi K, Nakajima H, Kamijima R, Tarutani M, Benson JM, Elloso MM, Gutshall LL, Naso MF, Iwakura Y, DiGiovanni J, Sano S: Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. In: Journal of Immunology. Band 186, Nr. 7, April 2011, S. 4481–9, doi:10.4049/jimmunol.1000148, PMID 21346238.
- Krueger JG, Fretzin S, Suárez-Fariñas M, Haslett PA, Phipps KM, Cameron GS, McColm J, Katcherian A, Cueto I, White T, Banerjee S, Hoffman RW: IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. In: The Journal of Allergy and Clinical Immunology. Band 130, Nr. 1, Juli 2012, S. 145–54.e9, doi:10.1016/j.jaci.2012.04.024, PMID 22677045, PMC 3470466 (freier Volltext).
- Dombrowski Y, Schauber J: Cathelicidin LL-37: a defense molecule with a potential role in psoriasis pathogenesis. In: Experimental Dermatology. Band 21, Nr. 5, Mai 2012, S. 327–30, doi:10.1111/j.1600-0625.2012.01459.x, PMID 22509827.
- S. Nischwitz, H. Faber, P. G. Sämann, H. S. Domingues, G. Krishnamoorthy: Interferon β-1a reduces increased interleukin-16 levels in multiple sclerosis patients. In: Acta Neurologica Scandinavica. Band 130, Nr. 1, Juli 2014, ISSN 1600-0404, S. 46–52, doi:10.1111/ane.12215, PMID 24571587 (nih.gov [abgerufen am 13. September 2021]).
- Swardfager W, Winer DA, Herrmann N, Winer S, Lanctôt KL: Interleukin-17 in post-stroke neurodegeneration. In: Neuroscience and Biobehavioral Reviews. Band 37, Nr. 3, März 2013, S. 436–47, doi:10.1016/j.neubiorev.2013.01.021, PMID 23370232.
- Ortiz ML, Kumar V, Martner A, Mony S, Donthireddy L, Condamine T, Seykora J, Knight SC, Malietzis G, Lee GH, Moorghen M, Lenox B, Luetteke N, Celis E, Gabrilovich D: Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. In: The Journal of Experimental Medicine. Band 212, Nr. 3, März 2015, S. 351–67, doi:10.1084/jem.20140835, PMID 25667306, PMC 4354367 (freier Volltext).
- Carbonell F, Heimpel H, Kubanek B, Fliedner TM: Growth and cytogenetic characteristics of bone marrow colonies from patients with 5q-syndrome. In: Blood. Band 66, Nr. 2, August 1985, S. 463–5, doi:10.1182/blood.V66.2.463.463, PMID 4016279.
- Cleve H, Kirk RL, Gajdusek DC, Guiart J: On the distribution of the Gc variant Gc Aborigine in Melanesian populations; determination of Gc-types in sera from Tongariki Island, New Hebrides. In: Acta Genetica et Statistica Medica. Band 17, Nr. 6, 1967, S. 511–7, doi:10.1159/000152104, PMID 4168861.
- Seppälä M, Rönnberg L, Karonen SL, Kauppila A: Micronized oral progesterone increases the circulating level of endometrial secretory PP14/beta-lactoglobulin homologue. In: Human Reproduction. Band 2, Nr. 6, August 1987, S. 453–5, doi:10.1093/oxfordjournals.humrep.a136569, PMID 3312283.
- First in the world regulatory approval of Novartis' Cosentyx(TM) in Japan for both psoriasis and psoriatic arthritis. Novartis AG, 26. Dezember 2014, abgerufen am 12. März 2015 (englisch).
- Cosentyx (secukinumab) An overview of Cosentyx and why it is authorised in the EU. (PDF) EMA, abgerufen am 13. September 2021 (englisch).
- Judith Stewart: Cosentyx FDA Approval History. 28. Januar 2021, abgerufen am 13. September 2021 (englisch).
- Kenneth B. Gordon, Andrew Blauvelt, Kim A. Papp, Richard G. Langley, Thomas Luger: Phase 3 Trials of Ixekizumab in Moderate-to-Severe Plaque Psoriasis. In: New England Journal of Medicine. Band 375, Nr. 4, 28. Juli 2016, ISSN 0028-4793, S. 345–356, doi:10.1056/NEJMoa1512711.
- Taltz ixekizumab. European public assessment report. EMA, abgerufen am 13. September 2021 (englisch).
- Brigitte M. Gensthaler: Neuer Antikörper für Psoriasis-Patienten zugelassen. In: Pharmazeutische Zeitung. 26. August 2021, abgerufen am 13. September 2021.
- Richard B. Warren, Andrew Blauvelt, Jerry Bagel, Kim A. Papp, Paul Yamauchi: Bimekizumab versus Adalimumab in Plaque Psoriasis. In: New England Journal of Medicine. Band 385, Nr. 2, 8. Juli 2021, ISSN 0028-4793, S. 130–141, doi:10.1056/NEJMoa2102388.
- Philip J. Mease, Mark C. Genovese, Maria W. Greenwald, Christopher T. Ritchlin, André D. Beaulieu: Brodalumab, an Anti-IL17RA Monoclonal Antibody, in Psoriatic Arthritis. In: New England Journal of Medicine. Band 370, Nr. 24, 12. Juni 2014, ISSN 0028-4793, S. 2295–2306, doi:10.1056/NEJMoa1315231.
- Kyntheum brodalumab. European public assessment report. EMA, 8. März 2017, abgerufen am 13. September 2021 (englisch).
- Leonardi CL, Kimball AB, Papp KA, Yeilding N, Guzzo C, Wang Y, Li S, Dooley LT, Gordon KB: Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). In: Lancet. Band 371, Nr. 9625, Mai 2008, S. 1665–74, doi:10.1016/S0140-6736(08)60725-4, PMID 18486739.