Hornhautdystrophie
Die Hornhautdystrophie (HD) ist eine Gruppe unterschiedlicher, angeborener nicht-entzündlicher, progredienter Erkrankungen (Dystrophien), die auf die Hornhaut beidseits beschränkt sind.[1]
| Klassifikation nach ICD-10 | |
|---|---|
| H18.5 | Hereditäre Hornhautdystrophien |
| ICD-10 online (WHO-Version 2019) | |
Die Erstbeschreibung einer Hornhautdystrophie stammt aus dem Jahre 1890 durch den deutschen Augenarzt Arthur Groenouw.[2]
Verbreitung
Die Häufigkeit einzelner Formen ist unterschiedlich, insgesamt jedoch selten.[1]
Im Rahmen von Syndromen
Bei folgenden Syndromen ist die Hornhautdystrophie ein wesentliches Krankheitsmerkmal:[3]
Ursache
Je nach genetischer Störung erfolgt die Vererbung autosomal-dominant, autosomal-rezessiv oder X-chromosomal-rezessiv.
Ursache der verschiedenen Hornhautdystrophien sind Mutationen in den Genen CHST6 (Genort 16q22), COL8A2 (1p34.3-p32.3), KRT3 (12q13), KRT12 (17q12), PIP5K3 (2q35), SLC4A11 (20p13-p12), TACSTD2 (1p32), TCF4 (18q21.2), TGFBI (5q31) und UBIAD1 (1p36.3).
Einteilung
Klinisch kann eine Einteilung nach Lokalisation erfolgen:[4]
Hinzu kommt die Autosomal-dominante Keratitis.[8]
Klassifikation
Im Jahre 2015 wurde die „IC3D Classification of Corneal Dystrophies—Edition 2“ veröffentlicht mit nachstehender Einteilung in vier Gruppen:[9]
- Epitheliale und subepitheliale Dystrophien
- Epitheliale Basalmembrandystrophie (EBMD)
- Epitheliale rezidivierende Erosionsdystrophie (ERED)
- Subepitheliale muzinöse HD (SMCD)
- Meesmann-HD (MECD), (Meesmann-Wilke-Syndrom)
- Lisch epitheliale HD (LECD)
- Gelatinöse tropfenförmige HD (GDLD)
- Dystrophien der Bowman-Lamelle
- Reis-Bücklers-HD (RBCD), (Granuläre Hornhautdystrophie Typ III)
- Thiel-Behnke HD (TBCD)
- Gittrige HDen (LCD;) – Typ 1 Varianten (III, IIIA, I/IIIA, IV)
- Granuläre HD Typ 1 (GCD1), (Groenouw I; klassische Form)
- Granuläre HD Typ 2 (GCD2), (Avellino-Dystrophie)
- Dystrophien des Stromas
- Makuläre HD (MCD), (Groenouw II; Fehr-Syndrom)
- Schnyder-HD (SCD)
- Kongenitale stromale HD (CSCD)
- Fleckchen HD (FCD)
- Posteriore amorphe HD (PACD)
- Zentral-wolkenförmige HD-François (CCDF)
- Prä-Descemet-HD (PDCD);
- Endotheliale Dystrophien
- Fuchs-endotheliale HD (FECD)
- Hintere polymorphe HD (PPCD)
- Kongenitale hereditäre Endotheldystrophie (CHED), (vormals CHED1 + 2)
- X-gebundene Endothel-Hornhautdystrophie (XECD)
Klinische Erscheinungen
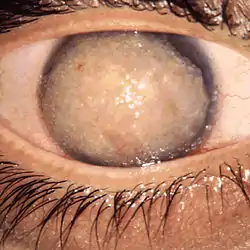
Klinische Kriterien sind Hornhauttrübung in unterschiedlicher Form und Ausprägung mit entsprechender Beeinträchtigung der Sehschärfe.[1]
Diagnose
Insbesondere bei beidseitigen Hornhauttrübungen sollte immer an Hornhautdystrophien gedacht werden. Die Diagnose stützt sich auf das Alter bei Erkrankungsbeginn und das Aussehen bei der Hornhaut bei der Spaltlampenuntersuchung.[1]
Differentialdiagnose
Abzugrenzen sind angeborene systemische Stoffwechselerkrankungen mit Hornhautbefall:[1]
Einzelnachweise
- Hornhautdystrophie. In: Orphanet (Datenbank für seltene Krankheiten).
- A. Groenouw: Knötchenförmige Hornhauttrübungen. (Noduli corneae) In: Archiv für Augenheilkunde. 1890, Bd. 21, S. 281–289.
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- W. Lisch, B. Seitz: Neue internationale Klassifikation der Hornhautdystrophien. In: Der Ophthalmologe. Bd. 108, 2011, S. 883, doi:10.1007/s00347-011-2388-8
- Hornhautdystrophie, superfizielle. In: Orphanet (Datenbank für seltene Krankheiten).
- Hornhautdystrophie, stromale. In: Orphanet (Datenbank für seltene Krankheiten).
- Hornhautdystrophie, posteriore. In: Orphanet (Datenbank für seltene Krankheiten).
- Keratitis, autosomal-dominante. In: Orphanet (Datenbank für seltene Krankheiten).
- J. Weiss, H. Møller, W. Lisch, S. Kinoshita, A. Aldave, M. Belin, T. Kivelä, M. Busin, F. Munier, B. Seitz, J. Sutphin, C. Bredrup, M. Mannis, C. Rapuano, G. Rij, E. Kim, G. Klintworth: IC³D-Klassifikation von Hornhautdystrophien. In: Klinische Monatsblätter für Augenheilkunde. Bd. 228, 2011, S. S1, doi:10.1055/s-0029-1245895.