Edman-Abbau
Der Edman-Abbau ist eine von dem schwedischen Biochemiker Pehr Edman im Jahr 1949 entwickelte Methode zur Proteinsequenzierung. Die Sequenzierung erfolgt dabei – im Gegensatz zum Schlack-Kumpf-Abbau – ausgehend vom N-terminalen Ende des Proteins.
Geschichte
Vor der Entwicklung des Edman-Abbaus wurden Proteine durch N-terminale Markierung mit 1-Fluor-2,4-dinitrobenzol (nach Frederick Sanger) versehen, anschließend wurden die Peptidbindungen der Proteine hydrolysiert, wodurch die N-terminale Aminosäure anhand der Markierung bestimmt wurde. Das Dinitrofluorbenzol wurde später gegen Dansylchlorid ersetzt, welches durch fluoreszente Derivate eine höhere Empfindlichkeit der Methode ermöglichte. Eine Bestimmung der Aminosäuresequenz konnte nur ungenau durch einen Zeitverlauf der Aminosäurefreisetzung nach Zugabe einer Exopeptidase zu einem Protein erreicht werden. Bei dem Edman-Abbau bleibt im Gegensatz zu den vorigen Methoden nach einer Abspaltung des Derivats der Rest des Proteins erhalten und kann in folgenden Zyklen zur Bestimmung der darauf folgenden Aminosäuren eingesetzt werden.
Heutzutage wird der Edman-Abbau nicht mehr häufig verwendet. Um die Aminosäuresequenz eines Proteins zu bestimmen, wird meist entweder die Gensequenz der DNA aus einer DNA-Sequenzierung oder aus einer Datenbank sequenzierter Genome wie tBLAST in silico in eine Proteinsequenz übersetzt. Durch molekulare Displays kann die Gensequenz eines Proteins erhalten werden. Um ein Protein direkt zu identifizieren (Ansequenzierung), können identifizierbare Teile eines Proteins neben dem Edman-Abbau auch per Massenspektrometrie untersucht werden. Die gemessene Masse eines Peptidfragments wird mit allen Peptid-Massen verglichen, die aus dem Genom berechnet werden können oder in einer Datenbank wie Mascot vorkommen. Zusammen mit einer gleichzeitigen Massenbestimmung aller Proteine mithilfe von MALDI-TOF kann so das gesamte Proteom zu einem bestimmten Zeitpunkt bestimmt werden.
Prinzip
Der Edman-Abbau erlaubt die Bestimmung der Reihenfolge von Aminosäuren (die Aminosäuresequenz) in einem Peptid durch wiederholte Endgruppenbestimmung.[1] Die Peptidkette wird dabei schrittweise abgebaut. Diese Reaktion wird zur Identifikation der N-terminalen Aminosäure eines Peptids genutzt und besteht aus der Umsetzung eines Peptids mit Phenylisothiocyanat, das auch als Edman-Reagenz bekannt ist.

Da jede Aminosäure einen unterschiedlichen Rest R besitzt, bildet jede ein unterschiedliches Phenylthiohydantoin-Derivat (PTH-Derivat). Die Identifikation der als PTH-Derivat abgespaltenen N-terminalen Aminosäure erfolgt chromatographisch durch Vergleich mit einem Standard. Man kann nacheinander weitere Abbaue an ein und demselben Protein (jeweils vorher verkürzt um eine Aminosäure am N-terminalen Ende) durchführen und so die Aminosäuresequenz nach und nach bestimmen. Ein Sequenator ist ein automatisiertes Gerät, das die unbeaufsichtigte Durchführung von bis zu 50 Abbaucyclen erlaubt.[2] Da die Reaktion mit einer relativen Ausbeute von > 98 % verläuft, nehmen einerseits die verschleppten Derivate aus vorhergehenden Zyklen und andererseits die unerwünschten Abspaltungsprodukte von Proteinen, die einen oder mehrere Abspaltungszyklus ausgesetzt haben, mit jedem Zyklus zu, so dass nach maximal 50 Zyklen das Signal-Rausch-Verhältnis unleserlich wird. Ein Zyklus dauert, je nach Variante, ein bis drei Stunden.
Mechanismus
Im hier dargestellten, von Zerong Wang[3] vorgeschlagenen Mechanismus bezeichnet R einen Alkylrest oder Wasserstoff, Ar bezeichnet einen Arylrest (meist einen Phenylrest) und die geschlängelte Linie steht für eine beliebig lange Fortsetzung der Proteinkette:
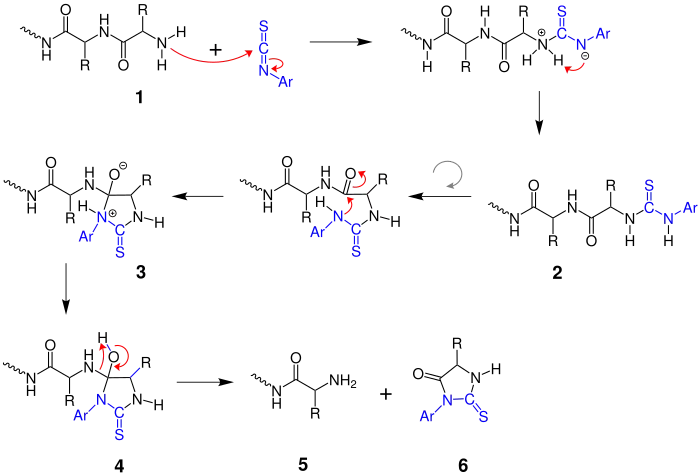
Zunächst versetzt man das zu analysierende Protein 1 mit einem Phenylisothiocyanatderivat. Dadurch erhält man über eine Zwischenstufe das Thioamid 2. Über eine „Biegung“ kommt es nun zu einem intramolekularen, nucleophilen Angriff, welcher eine Cyclisierung zum Anilinothiazolinon (ATZ-Derivat) zur Folge hat. Dadurch bildet sich das Zwitterion 3 aus welcher dann über einen Protonentransfer die heterocyclische Verbindung 4 entsteht. Dieses trägt noch die verbleibende Proteinkette, welche jedoch im folgenden Schritt abgespalten wird. Man erhält als Produkt das verbleibende Protein 5 und das Hydantoinderivat 6.
Dieser Abspaltungszyklus lässt sich nun mit dem Restpeptid mehrfach wiederholen. Mittels Chromatographie lassen sich nun das Hydantoinderivat 6 und somit die AS-Sequenz bestimmen.[4] Allerdings sind nur maximal 50 Zyklen[3] möglich. Rückstände – z. B. Reste des Hydantoinderivates aus vorherigen Schritten – können die Lösung so verunreinigen, dass nicht mehr nachvollziehbar ist, welche von ihnen aus dem aktuellen Zyklus abgespalten wurden oder welche von ihnen aus vorherigen Zyklen stammen. Dadurch ist eine Sequenzbestimmung nicht mehr sauber durchzuführen. Diesen Nachteil kann man umgehen, indem man die Peptid-Kette durch Proteolyse oder Bromcyanspaltung in mehrere sich überschneidende Teilketten zerlegt, bevor man mit der Sequenzierung beginnt.[3] Eine Spaltung ist auch bei Proteinen notwendig, deren N-Terminus modifiziert ist, z. B. N-terminal acetylierte Proteine oder Proteine mit N-terminaler Pyroglutaminsäure. Weitere Nachteile des Edman-Abbaus sind die hohen Kosten, die niedrige Sensitivität von etwa einem Picomol sowie eine Zykluszeit von etwa drei Stunden (mit der Zyklisierung als geschwindigkeitsbestimmenden Schritt).[4]
Modifikationen
Um die Nachteile eines Verlustes an Probenmaterial während der Extraktionen des Edman-Abbaus zu umgehen (engl. wash-out ‚Herauswaschen‘), wurde er so modifiziert, dass man ihn in fester Phase ablaufen lassen kann. Man spricht auch von „solid-phase support synthesis“ oder kurz SPSS. Auf diese Weise werden kurze Proteine oder Peptide automatisch sequenziert.[5] Ebenso werden auf einer PVDF-Membran immobilisierte Proteine aus einem Western Blot verwendet, sofern nur Blockierungslösungen ohne Proteine verwendet wurden. Durch eine Verwendung von 4-N,N-Dimethylaminoazobenzen-4'-Isothiocyanat (DABITC) sind farbige Aminosäurederivate erhältlich, was eine Analyse per Dünnschichtchromatographie erleichtert.[6] Durch eine Verwendung von 4-(1'-Cyanoisoindolyl)-phenylisothiocyanat können auch phosphorylierte Aminosäuren nachgewiesen werden.[7]
Außerdem hat der Edman-Abbau eine Anwendung in der Synthese von 2-Iminohydantoinen gefunden. Diese Reaktion soll hier exemplarisch mit der Reaktion von L-Leucinamid und 2-Bromphenyl-isothiocyanat dargestellt werden. Bei dieser Synthese können Reinheiten des Produktes von bis zu 99 % erreicht werden.[8]

Einzelnachweise
- P. Edman: A method for the determination of amino acid sequence in peptides. In: Arch. Biochem. Band 22, Nr. 2, 1949, S. 475, PMID 18134557.
- Paula Yurkanis Bruice: Organic Chemistry. 5. Auflage. Pearson Education, 2007, ISBN 978-3-8273-7190-4, S. 1200–1201.
- Z. Wang (Hrsg.): Comprehensive Organic Name Reactions and Reagents. 3 Volume Set. John Wiley & Sons, Hoboken, NJ, 2009, ISBN 978-0-471-70450-8, S. 954–955.
- Gavin E. Reid, Shane E. Tichy, James Pérez, Richard A. J. O’Hair, Richard J. Simpson: N-Terminal Derivatization and Fragmentation of Neutral Peptides via Ion—Molecule Reactions with Acylium Ions: Toward Gas-Phase Edman Degradation? In: J. Am. Chem. Soc. Nr. 123, 2001, S. 1184–1192, doi:10.1021/ja003070e.
- Richard A. Laursen: A Solid-State Edman Degradation. In: J. Am. Chem. Soc. Nr. 88, 1966, S. 5344–5346, doi:10.1021/ja00974a069.
- J. Y. Chang, E. H. Creaser: A novel manual method for protein-sequence analysis. In: Biochem J. 157, Nr. 1, 1976, S. 77–85, PMID 822842, PMC 1163818 (freier Volltext).
- Takayuki Shibata, Moses N. Wainaina, Takayuki Miyoshi, Tsutomu Kabashima, Masaaki Kai: A manual sequence method of peptides and phosphopeptides using 4-(1'-cyanoisoindolyl)phenylisothiocyanate. In: Journal of Chromatography A. 1218, Nr. 24, 2011, S. 3757–3762, doi:10.1016/j.chroma.2011.04.040, PMID 21531425.
- Ghotas Evindar, Robert A. Batey: Peptide Heterocycle Conjugates: A Diverted Edman Degradation Protocol for the Synthesis of N-Terminal 2-Iminohydantoins. In: Org. Lett. Band 5, Nr. 8, 2003, S. 1201–1204, doi:10.1021/ol034032d.