BLAST-Algorithmus
BLAST (Abkürzung für englisch Basic Local Alignment Search Tool) ist der Überbegriff für eine Sammlung der weltweit am meisten genutzten Programme zur Analyse biologischer Sequenzdaten. BLAST wird dazu verwendet, experimentell ermittelte DNA- oder Protein-Sequenzen mit bereits in einer Datenbank vorhandenen Sequenzen zu vergleichen. Als Ergebnis liefert das Programm eine Reihe lokaler Alignments, d. h. Gegenüberstellungen von Stücken der gesuchten Sequenz mit ähnlichen Stücken aus der Datenbank. Darüber hinaus gibt BLAST an, wie signifikant die gefundenen Treffer sind. Die Suche in der Datenbank erfolgt entweder über eine Webschnittstelle oder mit Hilfe von verschiedenen Stand-Alone-Programmen, die lokal installiert werden können.
Das Programm BLAST wurde von Stephen Altschul, Warren Gish, David J. Lipman, Webb Miller und Eugene Myers an den National Institutes of Health entwickelt.[1][2] Beteiligt an der Algorithmenentwicklung war auch Samuel Karlin.
Funktionsweise
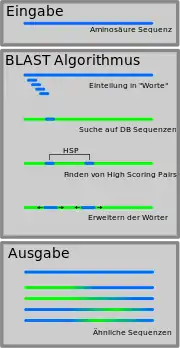
Die Idee des Algorithmus basiert auf der Wahrscheinlichkeit, dass Alignments mit vielen Treffern kurze Stücke von großer Identität besitzen. Diese Teilstücke werden dann während der Suche nach besseren und längeren Alignments weiter vergrößert.
Indem diese Segmente kurz gehalten werden, ist es möglich, die Abfragesequenz vor einer Suche zu bearbeiten und eine Tabelle aller möglichen Teilstücke mit ihrem Ursprung in der Originalsequenz vorzuhalten.
Dabei stellt der Algorithmus eine Liste aller benachbarten Worte fester Länge auf, die einen Treffer auf der Abfragesequenz mit einem höheren Scoring als ein zu wählender Parameter erzeugen würden. Anschließend wird die Zieldatenbank nach Worten in dieser Liste abgefragt und die gefundenen Treffer erweitert, um mögliche maximale zusammenhängende Treffer in beiden Richtungen zu finden.
Die Hauptanwendung von BLAST ist die Suche nach paralogen und orthologen Genen und Proteinen innerhalb eines oder mehrerer Organismen.
Methoden (Auswahl)
| Methode | Beschreibung |
|---|---|
| blastp | Vergleicht eine Aminosäuresequenz gegen eine Proteinsequenzdatenbank |
| PSI-BLAST | Position-Specific Iterative BLAST: Benutzt man, um entfernte Verwandte eines Proteins zu bestimmen.
Zuerst wird eine Liste aller sehr ähnlichen Proteine erstellt. Über diesen Proteinen wird ein Profil erstellt, eine Art gemittelte Sequenz. Daraufhin sendet man mit diesem Profil erneut eine Suchanfrage an die Proteindatenbank und erhält eine größere Gruppe ähnlicher Sequenzen. Mit dieser Gruppe kann man wieder ein neues Profil erstellen und den Prozess beliebig oft wiederholen. Dadurch, dass verwandte Proteine in die Suche miteinbezogen werden, ist PSI-BLAST viel empfindlicher bei der Ermittlung weit entfernter Verwandtschaften als das gewöhnliche Protein-Protein BLAST. |
| blastn | Vergleicht eine Nukleotidsequenz gegen eine Nukleotidsequenzdatenbank |
| blastx | Vergleicht eine Nukleotidsequenz (in allen Leserastern translatiert) gegen eine Proteindatenbank
Man kann diese Möglichkeit nutzen, um eine mögliche Translation einer bekannten Nukleotidsequenz zu finden. |
| tblastn | Vergleicht eine Proteinsequenz gegen eine Nukleotiddatenbank (dynamisch in allen Leserastern translatiert) |
| tblastx | Vergleicht die six-frame-Translation einer Nukleotidsequenz gegen die six-frame-Translationen einer Nukleotidsequenzdatenbank.
tblastx kann nicht mit der Nukleotiddatenbank auf der BLAST Webseite verwendet werden, da sie technisch sehr aufwändig ist! |
| megablast | megablast wird empfohlen zur Suche von identischen Sequenzen zu einer eigenen Sequenz. megablast wurde speziell erstellt, um besonders lange Sequenzen mit vorhandenen Gegenstücken aus der Datenbank abzugleichen.
discontiguous megablast wird empfohlen zur Suche nach Übereinstimmungen zwischen Sequenzen, die verteilt vorliegen, z. B. von verschiedenen Organismen stammen, und eine niedrige Übereinstimmungsrate haben. |
| cdart | cdart sucht Sequenzen mit einer möglichst identischen Anordnung von Proteindomänen unter Zuhilfenahme der CDD (=conserved domain)-Datenbank (Import von Übereinstimmungen aus SMART und Pfam) und vergleicht sie mit dem gesuchten Protein und dessen Domänen. |
Suchergebnisse
Die Homologie der bearbeiteten Suchsequenz wird anhand von Score und E-Wert definiert.
Der Score ist eine quantitative Bewertung der Ähnlichkeit der Suchsequenz mit einer bekannten Sequenz (je höher, desto höher ist auch die Identität der Sequenzen).
Der E-Wert gibt die erwartete Anzahl der Hits an, deren Score mindestens so groß ist wie der beobachtete (je kleiner, desto besser).
Die Abkürzungen vor und innerhalb der Suchergebnisse bedeuten (Auswahl):
| GenBank | gi-number|gb|accession|locus |
| EMBL Data Library | gi-number|emb|accession|locus |
| DDBJ, DNA Database of Japan | gi-number|dbj|accession|locus |
| NCBI Reference Sequence | gi-number|ref|accession|locus |
| SWISS-PROT | gi-number|sp|accession|name |
| General database identifier | database|identifier |
| Local Sequence identifier | identifier |
Anm: Die gi-Nummer ist eine Abfolge von Ziffern, die einen Datenbankeintrag des NCBI markiert.
Siehe auch
Literatur
- Stephen F. Altschul, Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang, Zheng Zhang, Webb Miller, David J. Lipman: Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. In: Nucleic Acids Research. Bd. 25, Nr. 17, 1997, S. 3389–3402, doi:10.1093/nar/25.17.3389.
- Lewis Y. Geer, Michael Domrachev, David J. Lipman, Stephen H. Bryant: CDART: Protein Homology by Domain Architecture. In: Genome Research. Bd. 12, 2002, ISSN 1088-9051, S. 1619–1623, PMID 12368255, doi:10.1101/gr.278202.
- Ian Korf, Mark Yandell, Joseph Bedell: BLAST. (An essential Guide to the Basic Local Alignment Search Tool). O’Reilly, Beijing u. a. 2003, ISBN 0-596-00299-8.
- Scott McGinnis, Thomas L. Madden: BLAST: at the core of a powerful and diverse set of sequence analysis tools. In: Nucleic Acids Research. Bd. 32, Supplement 2, 2004, S. W20–W25, doi:10.1093/nar/gkh435.
- Clare Sansom: Database searching with DNA and protein sequences: an introduction. (PDF; 203 kB) In: Briefings in Bioinformatics. Bd. 1, Nr. 1, 2000, ISSN 1467-5463, S. 22–32, PMID 11466971, doi:10.1093/bib/1.1.22
Weblinks
- NCBI Blast – National Center for Biotechnology Information
- NCBI BLAST+ – European Bioinformatics Institute
- BLAT – University of California, Santa Cruz
- Das Mitrion-C Open Bio Project bietet eine Mitrion FPGA basierte spezielle Version von BLAST auf SourceForge.
Einzelnachweise
- Stephen F. Altschul, Warren Gish, Webb Miller, Eugene W. Myers, David J. Lipman: Basic local alignment search tool. In: Journal of Molecular Biology. Bd. 215, 1990, ISSN 0022-2836, S. 403–410, doi:10.1016/S0022-2836(05)80360-2.
- Sense from Sequences: Stephen F. Altschul on Bettering BLAST. In: sciencewatch.com. 2000, archiviert vom Original am 23. April 2008; abgerufen am 7. Juli 2016.