Alkinone
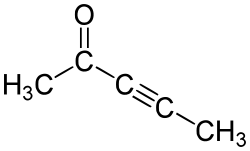
Alkinone sind chemische Verbindungen, die als funktionelle Gruppen eine C≡C-Dreifachbindung und eine nicht endständige Carbonylgruppe enthalten. Von besonderer Bedeutung sind diejenigen Verbindungen, bei denen beide Funktionalitäten direkt benachbart sind. Als konjugiertes System sind sie den Alkenonen verwandt und stellen wie diese einen aus drei Kohlenstoffatomen zusammengesetzten Synthesebaustein dar. Alkinone kann man aufgrund ihrer Reaktivität als Michael-System als Syntheseäquivalente von 1,3-Diketonen auffassen. Das nach dieser Systematik einfachste Alkinon ist Pent-3-in-2-on und damit dem Acetylaceton (Pentan-2,4-dion) analog. Formal ist But-3-in-2-on das einfachste Alkinon, das in der Reaktivität aber abweicht.
Herstellung
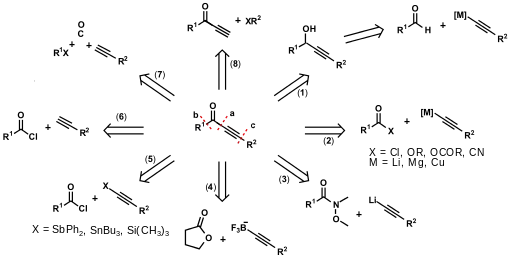
In den vergangenen Jahren wurden verschiedene Zugänge zu Alkinonen vorgestellt. Sie lassen sich nach der Art des retrosynthetischen Schnitts klassifizieren. Im Allgemeinen wird an der Stelle a geschnitten (Ansätze 1–6). Die Vorgehensweisen (1) und (2) stellen die klassischen Zugangsmöglichkeiten zu Alkinonen dar. Ausgehend von Lithiumacetyliden oder den äquivalenten Grignard-Reagenzien, ist es möglich zuerst den entsprechenden Propargylalkohol zu generieren, der dann zum gewünschten Produkt oxidiert wird (1). Nachteilig bei dieser Variante ist der benötigte Redoxschritt.[1][2][3]
Daher gehen die weiteren Syntheserouten von Carbonsäurederivaten aus. Die Ansätze (2) beschreiben die stöchiometrische Addition von Lithium-,[4] Magnesium-[5] oder Kupfer(I)-acetylide[6] an Carbonsäurederivate wie Säurechloride, Anhydride, Ester und Acylcyanide. Die Reaktion eines N-Methoxy-N-methyl-Weinreb-Amids mit einem Lithiumacetylid kann, wie in (3) beschrieben, zur Darstellung von Alkinonen verwendet werden,[7][8][9][10] ebenso Morpholinamide.[10] Einen ungewöhnlichen Weg zur Synthese von Alkinonen beschreibt die Route (4). Ein zuvor in stöchiometrischen Mengen dargestelltes Boralkinorganyl wird hierbei mit einem Lacton zu dem entsprechenden aliphatischen Alkinon, welches noch eine Hydroxygruppe trägt, umgesetzt.[11] Ansatz (5) stellt die Kreuzkupplungsreaktion von Zinn-[12] beziehungsweise Antimonacetyliden[13] mit Säurechloriden dar. Nachteilig ist, dass die Metallorganyle in einem vorgeschalteten Syntheseschritt separat dargestellt werden müssen. Alkinylsilane können auch in Anwesenheit von Lewis-Säuren wie AlCl3[14][15][16] oder Iod[17] mit Säurechloriden zur Reaktion gebracht werden. Mittlerweile gibt es hierzu auch katalytische Verfahren mit InBr3.[18]
Die Sonogashira-Hagihara-Reaktion eines Benzoesäurechlorids mit einem terminalen Alkin (6) in THF und in Gegenwart nur eines Äquivalents einer Aminbase, wurde im Arbeitskreis Müller entwickelt.[19] Die Ursprünge liegen jedoch über 30 Jahre zurück. 1977 fand Kenkichi Sonogashira heraus, dass sich die bereits bekannte Palladium(0)- und Kupfer(I)-katalysierte Sonogashira-Hagihara-Reaktion[20] auch auf Benzoesäurechloride und terminale Alkine als Substrate ausdehnen lässt.[21] Eine Methode, um gleichzeitig die Bindungen a und b zu knüpfen ist die carbonylierende Kupplung von Arylhalogeniden mit terminalen Alkinen (7). Gerade in Fällen, in denen die entsprechenden Säurechloride synthetisch schwer zu erhalten sind, ist diese Variante äußerst effizient. Die carbonylierende Kupplung war lange Zeit eine wenig genutzte Methode in der Synthese von Alkinonen. Insbesondere die hohen CO-Drücke, die für das Durchführen der Reaktion benötigt wurden, stellten ein Problem dar.[22][23][24] Im Jahr 2003 wurde ein kostengünstigeres Verfahren für die carbonylierende Alkinylierung unter CO-Normaldruck vorgestellt, bei dem Ammoniak als Base zum Einsatz kommt.[25]
Die Synthese über die Bindungsknüpfung an Stelle c wie in Beispiel (8), gehört zu den weniger verbreiteten Methoden zur Darstellung von Alkinonen. Hierbei bedient man sich wiederum einer Sonogashira-Hagihara-Reaktion. Es müssen jedoch Iodoniumsalze anstelle von Aryliodiden verwendet werden.[26]
Einzelnachweise
- D. Obrecht: Acid-Catalyzed Cyclization Reactions of Substituted Acetylenic Ketones: A new Approach for the Synthesis of 3-Halofurans, Flavones, and Styrylchromones, Helv. Chim. Acta, 1989, 72, S. 447–456 (doi:10.1002/hlca.19890720305).
- S. J. Pastine, D. Sames: Concise Synthesis of the Chemopreventitive Agent (±)-Deguelin via a Key 6-Endo Hydroarylation, Org. Lett., 2003, 5, S. 4053–4055 (doi:10.1021/ol035419j).
- A. L. K. S. Shun, E. T. Chernick, S. Eisler, R. R. Tykwinski: Synthesis of Unsymmetrically Substituted 1,3-Butadiynes and 1,3,5-Hexatriynes via Alkylidene Carbenoid Rearrangements, J. Org. Chem., 2003, 68, S. 1339–1347 (doi:10.1021/jo026481h).
- H. C. Brown, U. S. Racherla, S. M. Singh, Tetrahedron Lett., 1984, 25, S. 2411–2414.
- J. W. Kroeger, J. A. Nieuwland, J. Am. Chem. Soc., 1936, 58, S. 1861–1863.
- J. F. Normant: Organocopper(I) Compounds and Organocuprates in Synthesis, Synthesis, 1972, S. 63–80 (doi:10.1055/s-1972-21833).
- S. Nahm, S. M. Weinreb, Tetrahedron Lett., 1981, 22, S. 3815–3818.
- S. M. Bromidge, D. A. Entwistle, J. Goldstein, B. S. Orlek, Synth. Commun., 1993, 23, S. 487–494.
- T. L. Cupps, R. H. Boutin, H. Rapoport, J. Org. Chem., 1985, 50, S. 3972–3982.
- M. M. Jackson, C. Leverett, J. F. Toczko, J. C. Roberts, J. Org. Chem., 2002, 67, S. 5032–5035.
- J. Doubský, L. Streinz, L. Lešetický, B. Koutek, Synlett, 2003, 7, S. 937–942.
- M. W. Logue, K. Teng, J. Org. Chem., 1982, 47, S. 2549–2553.
- N. Kakusawa, K. Yamaguchi, J. Kurita, T. Tsuchiya, Tetrahedron Lett., 2000, 41, S. 4143–4146.
- L. Birkofer, A. Ritter, H. Uhlenbrauck, Chem. Ber., 1963, 96, S. 3280–3288.
- D. R. M. Walton, F. Waugh, J. Organomet. Chem., 1972, 37, S. 45–56.
- H. Newman, J. Org. Chem., 1973, 38, S. 2254–2255.
- J. S. Yadav, B. V. S. Reddy, M. S. Reddy, Synlett, 2003, 11, S. 1722–1724.
- J. S. Yadav, B. V. S. Reddy, M. S. Reddy, G. Parimala, Synthesis, 2003, S. 2390–2394.
- Karpov, A. S.; Müller, T. J. J., Org. Lett., 2003, 5, S. 3451–3454.
- K. Sonogashira, Y. Tohda, N. Hagihara, Tetrahedron Lett., 1975, 50, S. 4467–4470.
- Y. Tohda, K. Sonogashira, N. Hagihara, Synthesis, 1977, S. 777–778.
- T. Kobayashi, M. Tanaka, J. Chem. Soc., Chem. Commun., 1981, S. 333–334.
- K. Okuro, M. Furuune, M. Enna, M. Miura, M. Nomura, J. Org. Chem., 1993, 58, S. 4716–4721.
- L. Delude, A. M. Masdeu, H. Alper, Synthesis, 1994, S. 1149–1151.
- M. S. M. Ahmed, A. Mori, Org. Lett., 2003, 5, S. 3057–3060.
- U. Radharkrishnan, P. J. Stang, Org. Lett., 2001, 3, S. 859–860.