RNA-Seq
Als RNA-Seq, auch „Gesamt-Transkriptom-Shotgun-Sequenzierung“[1] genannt, wird die Bestimmung der Nukleotidabfolge der RNA bezeichnet, die auf Hochdurchsatzmethoden (Next-Generation Sequencing) basiert. Hierfür wird die RNA in cDNA übersetzt, damit die Methode der DNA-Sequenzierung angewendet werden kann. RNA-Seq enthüllt Informationen zur Genexpression, zum Beispiel wie unterschiedliche Allele eines Gens exprimiert werden und ermöglicht das Erkennen von posttranskriptionalen Modifikationen, sowie die Identifizierung von Fusions-Genen.[2]
RNA-Seq ist eine moderne sequenzbasierte Methode und basiert auf Sequenzierung der nächsten Generation (engl. next-generation sequencing). RNA-Seq hat klare Vorteile gegenüber den anderen Methoden. RNA-Seq hilft dabei, komplexe Transkriptome zu erforschen und gibt Aufschluss, welche Exons in der messenger-RNA zusammenfinden. Geringes Hintergrundrauschen, höhere Auflösung und hohe Reproduktionsraten in technischen als auch biologischen Replikaten sind klare Vorteile von RNA-Seq.[2]
Biologischer Hintergrund
Die Zelle verwendet nur einen Teil ihrer Gene. Darunter fallen die Haushaltsgene und die Gene der spezialisierten Zelle. Zum Beispiel haben Muskelzellen mechanische Eigenschaften, während β-Zellen der Langerhans-Inseln Insulin produzieren. Alle diese Zellen haben identische Gene, unterscheiden sich aber in ihrer Genexpression. Genexpression ist die Synthese von Proteinen aus der DNA. Die Genexpressionsanalyse oder auch Transkriptom-Analyse misst, welche Gene ein- oder ausgeschaltet sind. Wenn ein Gen angeschaltet ist, dann werden Teile des Gens in die mRNA übergeführt. Methoden der Genexpressionsanalyse, wie die des RNA-Seq, misst die Konzentration der mRNA in verschiedenen experimentellen Bedingungen (z. B. mit/ohne Medikamente). Die Genexpressionsanalyse folgt also der Frage, wie sich die mRNA-Konzentration durch Medikamente, in unterschiedlichen Entwicklungsstadien der Zelle, im gesunden oder erkrankten Zustand verhält.
Mit der RNA-Sequenz kann man den Mechanismus des alternativen Spleißens[3] sowie Fusionsgene[4] besser verstehen. Alternatives Spleißen ist der Prozess, bei dem die pre-RNA in verschiedene mRNAs und somit auch in verschiedene Proteine umgewandelt wird. Fusionsgene sind Hybridgene aus zwei vorher getrennten Genen, vereint in einem Gen. Fusionsgene entstehen durch Translokation, interstitielle Deletion oder durch chromosomale Inversion.
Arbeitsablauf
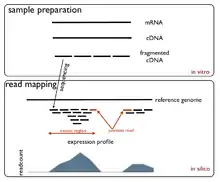
Probenaufbereitung
Meist interessiert man sich für die mRNA, die einen Entwurf für Proteine darstellt. Jedoch besteht die RNA einer Zelle zu 90 % aus rRNA. Um die mRNA von der rRNA zu trennen, gibt es standardisierte Methoden zur RNA-Reinigung, sogenannte „Ribosomal Depletion Kits“. Für die spätere Sequenzierung ist es notwendig, die mRNA zu fragmentieren, da die Sequenzierungstechniken nur eine bestimmte Leselänge haben. Die Fragmentierung kann sowohl vor (RNA-Fragmentierung) als auch nach der Konvertierung in die cDNA (cDNA-Fragmentierung) erfolgen. Die cDNA-Fragmentierung erzielt bessere Ergebnisse am 5'-Ende, jedoch zeigt sich eine schlechte Qualität in der Mitte des Transkripts, wo die RNA-Fragmentierung besser abschneidet.[2]
In der Probenaufbereitung muss man abwägen, ob man die Leserichtung, also strangspezifische Informationen, berücksichtigt. Damit kann man Artefakte ausschließen, die von der aRNA stammen. Jedoch ist das ein sehr zeit- und arbeitsintensiver Schritt.[5] Die aRNA inhibitiert durch Basenpaarung mit der komplementären mRNA deren Translation in der Zelle und beeinflusst die Genexpression einzelner Gene.
Sequenzierung
Es gibt mittlerweile sehr viele Hochdurchsatzmethoden, welche den Einbau eines einzelnen Nukleotids in die DNA in ein elektrisches Signal umwandeln. Viele dieser Methoden unterscheiden sich in der Durchführung. Hier nun ein Beispiel für die Sequenzierung auf dem Illumina Genome Analyzer II:[6]
- Fragmentierung der cDNA
- Reinigung, Reparatur der Fragmentenden
- Adapter werden an die Probe ligiert
- Die Proben werden mit einem Agarose-Gel nach ihrer Größe aufgetrennt
- PCR
- Reinigung und Sequenzierung
Read Mapping
Die wohl größte Herausforderung in der Datenanalyse von RNA-Seq besteht darin, die gelesenen Fragmente (Reads) dem Referenzgenom zuzuordnen. Das mag für einen einzelnen Read trivial erscheinen, jedoch für Millionen von Reads brauchen etablierte Alignmentverfahren wie z. B. BLAST 43 Stunden um 10 Millionen Reads mit einer Länge von 32 bp dem Referenzgenom zuzuordnen.[6] Deshalb war es notwendig, neue Algorithmen für das Read Mapping zu entwerfen.
Beim Read Mapping gibt es Algorithmen, die das Spleißen berücksichtigen, wie z. B. exon first oder seed and extend, sowie Algorithmen, die das Spleißen nicht berücksichtigen wie z. B. seed methods oder Burrows-Wheeler Aligner.[7]
Einzelnachweise
- Ryan D. Morin, Matthew Bainbridge, Anthony Fejes, Martin Hirst, Martin Krzywinski, Trevor J. Pugh, Helen McDonald, Richard Varhol, Steven J.M. Jones, and Marco A. Marra.: Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. In: BioTechniques. 45, Nr. 1, 2008, S. 81–94. doi:10.2144/000112900. PMID 18611170.
- Zhong Wang, Mark Gerstein, Michael Snyder: RNA-Seq: a revolutionary tool for transcriptomics. In: Nature Reviews Genetics. 10, Nr. 1, Januar 2009, S. 57–63. doi:10.1038/nrg2484. PMID 19015660. PMC 2949280 (freier Volltext).
- Trapnell C, Pachter L, Salzberg SL: TopHat: discovering splice junctions with RNA-Seq.. In: Bioinformatics. 25, Nr. 9, 2009, S. 1105–1111. doi:10.1093/bioinformatics/btp120. PMID 19289445. PMC PMC2672628 (freier Volltext).
- Teixeira MR: Recurrent fusion oncogenes in carcinomas.. In: Crit Rev Oncog. 12, Nr. 3–4, 2006, S. 257–271. PMID 17425505.
- Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK et al.: Stem cell transcriptome profiling via massive-scale mRNA sequencing.. In: Nat Methods. 5, Nr. 7, 2008, S. 613–619. doi:10.1038/nmeth.1223. PMID 18516046.
- Wilhelm BT, Landry JR: RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing.. In: Methods. 48, Nr. 3, 2009, S. 249–257. doi:10.1016/j.ymeth.2009.03.016. PMID 19336255.
- Garber M, Grabherr MG, Guttman M, Trapnell C: Computational methods for transcriptome annotation and quantification using RNA-seq.. In: Nat Methods. 8, Nr. 6, 2011, S. 469–477. doi:10.1038/nmeth.1613. PMID 21623353.