Meyer-Schuster-Umlagerung
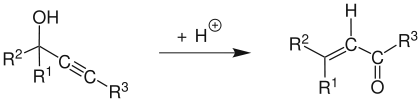
Die Meyer-Schuster-Umlagerung ist eine chemische Reaktion, bei der sich säurekatalysiert sekundäre und tertiäre Propargylalkohole (R1= H oder Organylgruppe, R2= H oder Organylgruppe, R3= H oder Organylgruppe) zu α,β-ungesättigten Ketonen umlagern. Bei endständiger Alkingruppe bilden sich α,β-ungesättigte Aldehyde. Die Namensreaktion wurde von Kurt H. Meyer und Kurt Schuster 1922 entdeckt und publiziert.[1]
Es wurden mehrere Übersichtsartikel zur Meyer-Schuster-Umlagerung veröffentlicht.[2][3][4]
Die basenkatalysierte Variante der Umlagerung ist als Faworski-Reaktion bekannt.
Mechanismus
Der Reaktionsmechanismus[5] beginnt mit der Protonierung des Alkohols, wobei Wasser in einer E1 Reaktion eliminiert wird und das Allen aus dem Alkin gebildet wird. Durch Angriff des Wassermoleküls auf das Carbokation und anschließender Deprotonierung gefolgt von einer Tautomerie bildet sich eine α,β-ungesättigte Carbonyl-Verbindung.[6]
Der Reaktionsmechanismus wurde von Edens et al. untersucht.[7] Sie fanden drei charakteristische Schritte: (1) die schnelle Protonierung des Sauerstoffs, (2) die langsame, geschwindigkeitsbestimmende Schritt der sigmatropen 1,3-Umlagerung der protonierten Hydroxygruppe und die Keto-Enol-Tautomerie, gefolgt von der schnellen Deprotonierung.
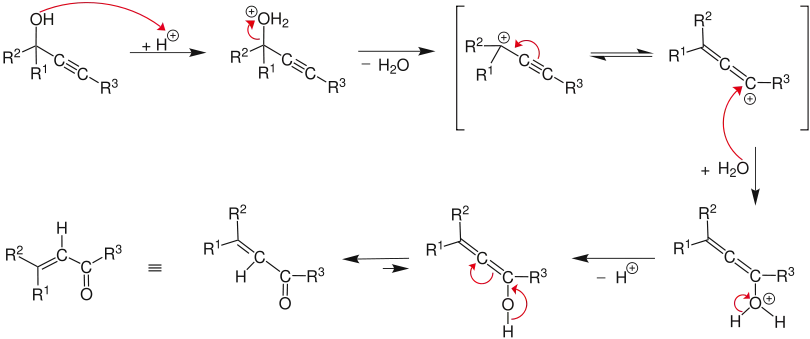
In einer Untersuchung des geschwindigkeitsbestimmenden Schritts der Meyer-Schuster-Umlagerung zeigten Andres et al., dass die treibende Kraft der Reaktion die irreversible Bildung der ungesättigten Carbonyl-Verbindung über das Carbonium-Ion ist.[8] Sie zeigten auch, dass die Reaktion durch das verwendete Lösungsmittel unterstützt wird. Dies wurde eingehender durch Tapi et al. untersucht, die zeigten, dass die Bildung von Lösungsmittelkäfigen den Übergangszustand stabilisiert.[9]
Rupe-Umlagerung
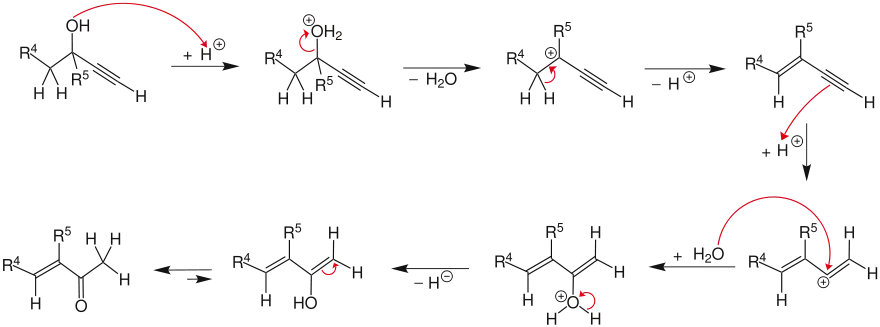
Die Reaktion von tertiären Alkoholen (R4,R5= Organylgruppe), die in α-Stellung eine Alkingruppe enthalten, führt nicht zu den erwarteten Alkoholen, sondern zu α,β-ungesättigten Ketonen über eine Enin-Zwischenstufe.[10][11] Diese Reaktion erfolgt für tertiäre Alkohole in Konkurrenz zur Meyer-Schuster-Umlagerung und wird als Rupe-Umlagerung bezeichnet (nach Hans Rupe). Im ersten Schritt findet eine Protonierung des Alkohols statt, wobei Wasser abgespalten wird. Dabei entsteht ein Propargylation, welches deprotoniert wird und zu einem Enin reagiert. Nach Protonierung dieser Zwischenstufe und der Reaktion mit Wasser entsteht unter Tautomerization das α,β-ungesättigte Keton.[12]
Katalysatoren
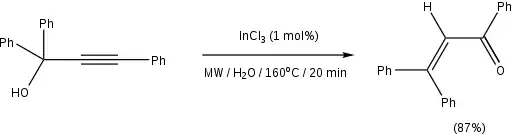
Die Bedingungen der traditionellen Meyer-Schuster-Umlagerung unter Verwendung von starken Säuren als Katalysator führt im Falle von tertiären Alkoholen zu Nebenreaktionen wie der Rupe-Umlagerung.[2] Durch die Verwendung von Übergangsmetall- und Lewis-Säuren als Katalysatoren lässt sich die Reaktion unter milderen Bedingungen durchführen, zum Beispiel durch Ruthenium-[13] und Silber-haltige[14] Katalysatoren. Carieno et al. berichteten über die Verwendung von Mikrowellenbestrahlung mit InCl3 als Katalysator, die zu exzellenten Ausbeuten und kurzen Reaktionszeiten und bemerkenswerter Stereoselektivität führte.[15] Ein Beispiel aus der Veröffentlichung ist wie folgt:
Anwendungen
Die Meyer-Schuster-Umlagerung findet eine Reihe von Anwendungen, von der Umwandlung von ω-Alkin-ω-Carbinol Lactamen in Enamide mit PTSA als Katalysator[16] über die Synthese von α,β-ungesättigten Thioester aus γ-Schwefel substituierten Propargylalcoholen[17] bis zur Umlagerung von 3-Alkin-3-Hydroxy-1H-Isoindolen unter mild-sauren Bedingungen zu α,β-ungesättigten Carbonylverbindungen.[18] Eine der interessantesten Anwendungen ist die Synthese eines Teilstruktur von Paclitaxel in einer diastereoselektiven Synthese, die nur zum E-Alken führt.[19]
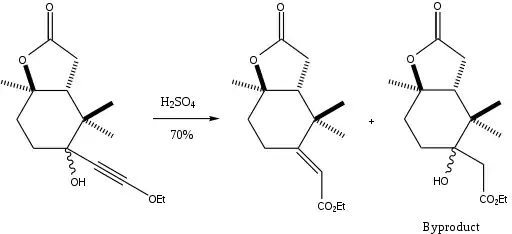
Der oben gezeigte Syntheseschritt erfolgt in 70%iger Ausbeute, und sogar in 91%iger Ausbeute, wenn das Beiprodukt in einem anderen Schritt in das Meyer-Schuster-Produkt umgewandelt wird.
Einzelnachweise
- Kurt H. Meyer, Kurt Schuster: Umlagerung tertiaerer Äthinyl-carbinole in ungesättigte Ketone. In: Berichte der deutschen chemischen Gesellschaft (A und B Serie). 55, 1922, S. 819–823, doi:10.1002/cber.19220550403.
- S. Swaminathan, K. V. Narayanan: Rupe and Meyer-Schuster rearrangements. In: Chemical Reviews. 71, 1971, S. 429–438, doi:10.1021/cr60273a001.
- S A Vartanyan, Sh O Babanyan: REARRANGEMENT OF ACETYLENIC COMPOUNDS WITH PARTICIPATION OF THE π-ELECTRONS OF THE TRIPLE BOND. In: Russian Chemical Reviews. 36, 1967, S. 670–686, doi:10.1070/RC1967v036n09ABEH001681.
- Douglas A. Engel, Gregory B. Dudley: The Meyer-Schuster rearrangement for the synthesis of α,β-unsaturated carbonyl compounds. In: Organic & Biomolecular Chemistry. 7, 2009, S. 4149, doi:10.1039/b912099h.
- Li, J.J. In Meyer-Schuster rearrangement; Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin, 2006; pp 380-381.(doi:10.1007/978-3-642-01053-8_159)
- László Kürti und Barbara Czakó: Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms, Elsevier Academic Press, 2005, S. 284–285, ISBN 978-0-12-429785-2.
- Edens, M.; Boerner, D.; Chase, C. R.; Nass, D.; Schiavelli, M. D. J. Org. Chem. 1977, 42, 3403-3408. (doi:10.1021/jo00441a017)
- Andres, J.; Cardenas, R.; Silla, E.; Tapia, O. J. Am. Chem. Soc. 1988, 110, 666-674. (doi:10.1021/ja00211a002)
- Tapia, O.; Lluch, J.M.; Cardena, R.; Andres, J. J. Am. Chem. Soc. 1989, 111, 829-835. (doi:10.1021/ja00185a007)
- Rupe, H.; Kambli, E. Helv. Chim. Acta 1926, 9, S. 672 (doi:10.1002/hlca.19260090185).
- Li, J.J. In Rupe rearrangement; Name Reactions: A Collection of Detailed Reaction Mechanisms; Springer: Berlin, 2006; S. 513-514(doi:10.1007/978-3-642-01053-8_224).
- László Kürti und Barbara Czakó: Strategic Applications of Named Reactions in Organic Synthesis: Background and Detailed Mechanisms, Elsevier Academic Press, 2005, ISBN 978-0-12-429785-2, S. 284–285.
- Cadierno, V.; Crochet, P.; Gimeno, J. Synlett 2008, 1105-1124. (doi:10.1055/s-2008-1072593)
- Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. J. Am. Chem. Soc. 2007, 129, 12902-12903. (doi:10.1021/ja074350y)
- Cadierno, V.; Francos, J.; Gimeno, J. Tetrahedron Lett. 2009, 50, 4773-4776.(doi:10.1016/j.tetlet.2009.06.040)
- Chihab-Eddine, A.; Daich, A.; Jilale, A.; Decroix, B. J. Heterocycl. Chem. 2000, 37, 1543-1548.(doi:10.1002/jhet.5570370622)
- Yoshimatsu, M.; Naito, M.; Kawahigashi, M.; Shimizu, H.; Kataoka, T. J. Org. Chem. 1995, 60, 4798-4802.(doi:10.1021/jo00120a024)
- Omar, E.A.; Tu, C.; Wigal, C.T.; Braun, L.L. J. Heterocycl. Chem. 1992, 29, 947-951.(doi:10.1002/jhet.5570290445)
- Crich, D.; Natarajan, S.; Crich, J.Z. Tetrahedron 1997, 53, 7139-7158.(doi:10.1016/S0040-4020(97)00411-0)