Ireland-Claisen-Umlagerung
Die Ireland-Claisen-Umlagerung ist eine Namensreaktion aus dem Bereich der Organischen Chemie und eine Variante der herkömmlichen Claisen-Umlagerung. Die Reaktion wurde um 1972 von dem amerikanischen Chemiker Robert E. Ireland (1933–2012) entwickelt[1] und stellt eine sehr milde [3,3]-sigmatrope Umlagerung von enolisierten und silylierten Allylestern dar. Ireland-Claisen-Umlagerungen führen zu γ,δ-ungesättigten Carbonsäuren und lassen sich je nach Konfiguration des Esterenolats mit ausgezeichneter Diastereoselektivität durchführen.

Übersichtsreaktion
Bei der Ireland-Claisen-Umlagerung wird zunächst aus einem Allylester (1) mit einer starken, nicht-nukleophilen Base (z. B. Lithiumdiisopropylamid, LDA) in einem geeigneten Lösungsmittel (z. B. Tetrahydrofuran, THF) bei −78 °C das entsprechende Ester-Enolat erzeugt, welches dann sofort mit Chlorsilanen (z. B. Trimethylsilylchlorid, (CH3)3SiCl) zu einem O-Allyl-O-silylketenacetal (2) umgesetzt wird. Beim Auftauen auf Raumtemperatur findet dann eine [3,3]-sigmatrope Umlagerung statt, nach dessen saurer Aufarbeitung man die entsprechende γ,δ-ungesättigte Carbonsäure (3) erhält:

Die Ireland-Claisen-Umlagerung führt je nach Stereochemie des Enolats und der Art der Substituenten R1–R3 zu diastereomeren Produkten, deren stereochemischer Verlauf im weiteren Verlauf des Artikels behandelt wird.
Im Vergleich zu der klassischen Claisen-Umlagerung verläuft die Ireland-Variante aufgrund der zusätzlichen Mesomeriestabilisierung des entstehenden Silyesters (vor der wässrigen Aufarbeitung) unter deutlich milderen Bedingungen ab.
Reaktionsmechanismus
Der ersten Schritt der Ireland-Claisen-Umlagerung ist die Herstellung des O-Allyl-O-silylketenacetals aus einem beliebigen Allylester. Dieser wird dazu zunächst am α-Kohlenstoffatom durch eine sterisch anspruchsvolle, nicht-nukleophile Base deprotoniert und das entsprechende Ester-Enolat gebildet, welches unmittelbar durch eine Silylierung abgefangen wird. Bringt man anschließend die Temperatur wieder auf Raumtemperatur, so findet die [3,3]-sigmatrope Umlagerung statt. Mechanistisch betrachtet verläuft diese über einen sechsgliedrigen Übergangszustand, der energetisch bevorzugt in der Sesselkonformation vorliegt. Dadurch entsteht zunächst ein α-allylierter Silylester, welcher im Allgemeinen so instabil ist, dass dieser direkt im Zuge einer sauren Aufarbeitung in die entsprechende γ,δ-ungesättigte Carbonsäure überführt wird.
Betrachtet man den einfachsten Allylester ohne Substituenten und damit ohne notwendige Berücksichtigung der Stereochemie, so ergibt sich folgender Mechanismus:

Das zuvor gebildete Silylketenacetal geht beim Auftauen auf Raumtemperatur eine [3,3]-sigmatrope Umlagerung ein, wobei der Übergangszustand sechsgliedrig verläuft. Die dabei neu gebildete C–C-Bindung ist in grün markiert.
Stereoselektivität
Präparativ bedeutender sind Ireland-Claisen-Umlagerungen mit kontrollierter Stereochemie. Die Stereoselektivität bei Ireland-Claisen-Umlagerungen ist die Folge von kinetischer Kontrolle, also die Konsequenz, dass die Umlagerung über den energieärmsten Übergangszustand erfolgt. Da der günstigste Übergangszustand zweckmäßig sechsgliedrig und sesselförmig ist, ist die Orientierung der Substituenten relativ zur Sesselstruktur entscheiden für die Stereoselektivität.
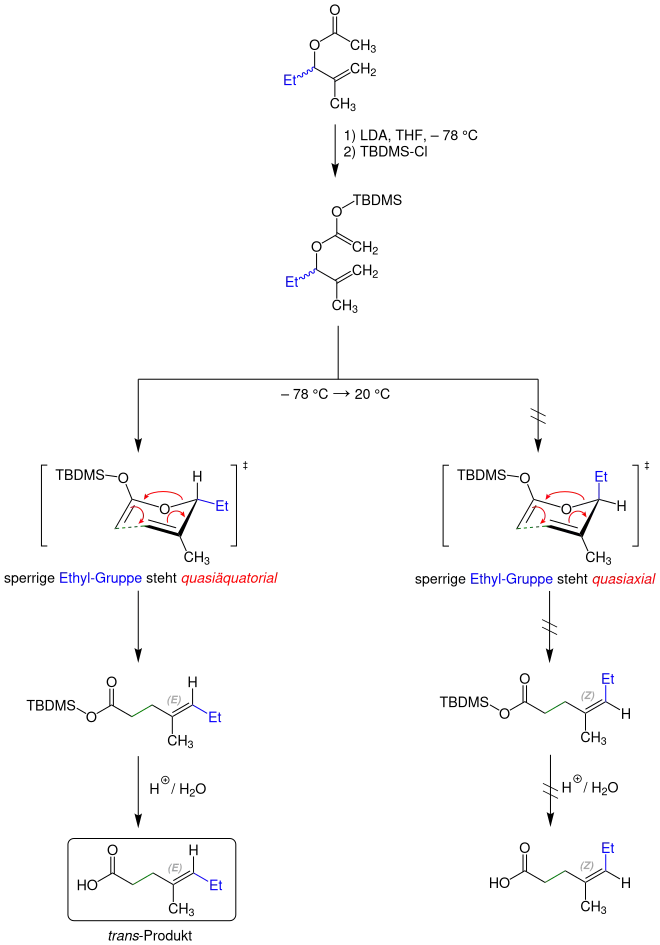
In dem obigen Beispiel wird ein Allylester, welcher ein Chiralitätszentrum am Allyl-Rest besitzt, in racemischer Form eingesetzt. Zunächst wird daraus das entsprechende O-Allyl-O-silylketenacetal gebildet (TBDMS-Cl steht für tert-Butyldimethylsilylchlorid, eine Verbindung zur Einführung einer tert-Butyldimethylsilyl-Schutzgruppe, welche neben einer TMS-Gruppe häufig zum Abfangen von Enolaten genutzt wird). Die Umlagerungsreaktion kann nun entweder über einen Übergangszustand erfolgen, in dem der sperrige Substituent (blau markierte Ethyl-Gruppe) quasiäquatorial oder quasiaxial angeordnet ist. Ersterer ist dabei kinetisch deutlich bevorzugter, da eine sterische Überfrachtung sowie 1,3-diaxiale Wechselwirkung minimiert wird. Der Reaktionspfad über den anderen Übergangszustand wird praktisch nicht bestritten. Somit wird am Ende der Reaktion nach wässriger Aufarbeitung ausschließlich das trans-Produkt (Doppelbindung ist E-konfiguriert) erhalten.
Diastereoselektivität
Eine weitere Möglichkeit, die Stereoselektivität bei der Ireland-Claisen-Umlagerung zu steuern, liegt in der Variation der Konfiguration der eingesetzten Enolate. Die Umlagerung verläuft dann in allen Fällen hoch diastereoselektiv:[2]

In dem gezeigten Beispiel wird von einem Propionsäureallylester, dessen Allyl-Rest E-konfiguriert ist, ausgegangen. Die Enolatisierung und Silylierung liefert in reinem THF ausschließlich das entsprechende E-Enolat im O-Allyl-O-silylketenacetal. Im Übergangszustand ist somit die Position der Methyl-Gruppe in α-Position festgelegt. Die Umlagerung führt nach wässriger Aufarbeitung zum anti-Diastereoisomer. Setzt man hingegen bei der Enolatisierung ein THF/DMPU-Gemisch als Lösungsmittel ein, so wird stereoselektiv das entsprechende Z-Enolat erzeugt. Über den entsprechenden festgelegten Übergangszustand wird schließlich das syn-Diastereoisomer erhalten. In beiden Endprodukten wurden dabei zwei neue, einander benachbarte Stereozentren eingeführt.
Literatur
- Reinhard Brückner: Reaktionsmechanismen – Organische Reaktionen, Stereochemie, Moderne Synthesemethoden. 3. Auflage. Springer Spektrum, Berlin-Heidelberg 2015, ISBN 978-3-662-45683-5.
Weblinks
Einzelnachweise
- Robert E. Ireland, Richard H. Mueller: Claisen rearrangement of allyl esters. In: J. Am. Chem. Soc.. 94, 1972, S. 5897–5898. doi:10.1021/ja00771a062.
- Robert E. Ireland, Peter Wipf, Joseph D. Armstrong III: Stereochemical control in the ester enolate Claisen rearrangement. 1. Stereoselectivity in silyl ketene acetal formation. In: J. Org. Chem.. 56, 1991, S. 650–657. doi:10.1021/jo00002a030.