Dihydropyrimidin-Dehydrogenase
Die Dihydropyrimidin-Dehydrogenase (DPD) ist ein Enzym aus der Gruppe der Oxidoreduktasen, das einen Schritt im Abbau von körpereigenen Pyrimidinen wie Uracil und Thymin katalysiert. Zusätzlich baut es verschiedene Zytostatika wie 5-Fluoruracil (5-FU) und Capecitabin ab. Es gibt angeborene DPD-Mangelzustände (DPD-Mangel, E79 nach ICD-10), die vor einer geplanten Therapie mit Fluorpyrimidinen mittels Labortests ausgeschlossen werden sollten. Falls ein solcher Mangelzustand vorliegt, müssen die entsprechenden Medikamente entweder in reduzierter Dosierung verabreicht werden oder dürfen (bei einem vollständigen Fehlen der DPD-Enzymaktivität) gar nicht gegeben werden.[1]
| Dihydropyrimidin-Dehydrogenase | ||
|---|---|---|
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 1022 Aminosäuren | |
| Sekundär- bis Quartärstruktur | Homodimer | |
| Kofaktor | FAD, FMN, (4Fe-4S) | |
| Bezeichner | ||
| Gen-Name | DPYD | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 1.3.1.2, Oxidoreduktase | |
| Reaktionsart | Redoxreaktion | |
| Substrat | 5,6-Dihydrouracil + NADP+ | |
| Produkte | Uracil + NADPH + H+ | |
| Vorkommen | ||
| Übergeordnetes Taxon | Eukaryoten, Bakterien | |
| Orthologe | ||
| Mensch | Hausmaus | |
| Entrez | 1806 | 99586 |
| Ensembl | ENSG00000188641 | ENSMUSG00000033308 |
| UniProt | Q12882 | Q8CHR6 |
| Refseq (mRNA) | NM_000110 | NM_170778 |
| Refseq (Protein) | NP_000101 | NP_740748 |
| Genlocus | Chr 1: 97.08 – 97.92 Mb | Chr 3: 118.56 – 119.43 Mb |
| PubMed-Suche | 1806 | 99586 |
Katalysierte Reaktionen
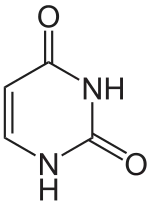 + NADPH/H+
+ NADPH/H+ 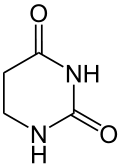 + NADP+
+ NADP+

Uracil wird zu Dihydrouracil hydriert.
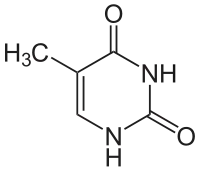 + NADPH/H+
+ NADPH/H+ 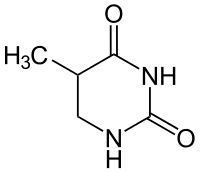 + NADP+
+ NADP+
Thymin wird zu 5,6-Dihydrothymin hydriert.
Die katalysierte Reaktion ist reversibel und geschwindigkeitsbestimmend, das heißt, je nach Verfügbarkeit der Ausgangs- und Endprodukte stellt sich ein Gleichgewicht ein. Das Enzym ist in allen Eukaryoten, so auch dem Schleimpilz Dictyostelium, und vielen Bakterien zu finden. Ein naher Verwandter in Pflanzen ist die Dihydroorotat-Reduktase.[2]
DPD-Mangelzustände und -Hemmstoffe
Das Enzym wird durch verschiedene Stoffe gehemmt, u. a.
- Bromvinyluracil, ein Abbauprodukt des Virostatikums Brivudin (deswegen kann die gleichzeitige Gabe von 5-FU bzw. seinen Vorläufern und Brivudin zu gefährlichen Interaktionen führen),
- Uracil,
- Eniluracil.
Etwa 9 Prozent der europäischen Bevölkerung weisen eine verminderte DPD-Enzymaktivität auf. Meist handelt es sich um eine relative Verminderung der Enzymaktivität. Ein absoluter Enzymmmangel, d. h. ein vollständiges Fehlen der Enzymaktivität ist sehr selten. Für Patienten, die eine Therapie mit Fluorpyrimidinen (5-FU, Capecitabin, Tegafur, aber auch das Antimykotikum 5-Fluorcytosin) erhalten sollen, sind diese Mangelzustände klinisch relevant. Werden die genannten Medikamente in den üblichen Dosierungen verabreicht, kann dies bei Personen mit DPD-Mangel zu mehr oder minder schweren toxischen Nebenwirkungen führen, da die Medikamente nur verzögert abgebaut werden und daher länger wirken. Bei dem sehr seltenen vollständigen DPD-Enzymmangel kann dies unter Umständen tödlich enden, da die Medikamente gar nicht abgebaut werden und daher sehr lange wirken. Den meisten Fällen von DPD-Mangel liegen erbliche Varianten (Polymorphismen) des Gens DPYD zugrunde. Im Jahr 2020 waren mehr als 2190 Genvarianten, die sich auf 160 verschiedene Allele von DPYD verteilten, bekannt. Die relative Häufigkeit (Prävalenz) dieser Genvarianten war im Allgemeinen relativ niedrig. Für die meisten Genvarianten war nicht bekannt, welchen Einfluss sie auf die DPD-Enzymaktivität hatten. Nur für eine Reihe von häufigeren Varianten gab es hinreichende klinische Evidenz, inwieweit sie die DPD-Enyzmaktivität reduzierten.[3]
Testung auf DPD-Mangel
Im März 2020 empfahl der Ausschuss für Risikobewertung im Bereich der Pharmakovigilanz (PRAC) bei der Europäischen Arzneimittel-Agentur (EMA) die grundsätzliche künftige Testung von Patienten, die eine Therapie mit 5-FU, Capecitabin oder Tegafur erhalten sollten, auf DPD-Enzymmangel. Am 30. April 2020 schloss sich die EMA dieser Empfehlung an und am 29. Juli 2020 folgte das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) mit einer entsprechenden Empfehlung.[1] Für den Fall des Nachweises eines DPD-Mangels wird eine Dosisreduktion der genannten Medikamente empfohlen. Bei vollständigem Fehlen der DPD-Enzymaktivität dürfen die Medikamente nicht gegeben werden.[4]
Im Falle einer genetischen Testung soll das Vorhandensein der folgenden vier DPYD-Varianten vor einer Therapie mit 5FU, Capecitabin oder Tegafur untersucht werden:[5]
- DPYD*2A (c.1905+1G>A; rs3918290, „Exon-14-skipping“)
- DPYD*13A (c.1679T>G; rs55886062)
- Polymorphismus c.2846A>T (rs67376798)
- Haplotyp B3 (rs75750017182, c.1129-5923C>G; rs56038477, c.1236G>A; rs56276561, c483+18G>A)
Ein DPD-Aktivitätsmangel lässt sich auch am Uracilspiegel im Serum ablesen. Ein Serumspiegel über 16 ng/ml Serum gilt als pathologisch. Die DPD-Aktivität lässt sich auch in Leukozyten messen. Im Ergebnis erhält man einen Aktivitätsscore:
- 2: normale Aktivität, geplante Dosis kann verabreicht werden
- 1 bis 1,5: Initialdosis auf 25–50 % reduzieren. Je nach Toxizität steigern.
- 0,5: DPD-Phänotypisierung. Bei Bestätigung 5-FU usw. vermeiden oder stark reduzieren
- 0: Keine Therapie mit 5-FU usw. Nur 0,5 % der europäischen Bevölkerung[6][7]
Aufgrund schwierigerer Standardisierbarkeit ist die Uracil-Spiegelmessung im Vergleich zur genetischen Testung derzeit nur wenig im Routinegebrauch.
Weblinks
Einzelnachweise
- Fluorouracil, Capecitabin, Tegafur und Flucytosin: Empfehlung zur Testung und Behandlung. Bundesinstitut für Arzneimittel und Medizinprodukte, 4. August 2020, abgerufen am 26. September 2020.
- Uniprot-Eintrag
- C. A. T. C. Lunenburg, C. H. van der Wouden, M. Nijenhuis, M. H. Crommentuijn-van Rhenen, N. J. de Boer-Veger, A. M. Buunk, E. J. F. Houwink, H. Mulder, G. A. Rongen, R. H. N. van Schaik, J. van der Weide, B. Wilffert, V. H. M. Deneer, J. J. Swen, H. J. Guchelaar: Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction of DPYD and fluoropyrimidines. In: Eur J Hum Genet. Band 28, Nr. 4, April 2020, S. 508–517, doi:10.1038/s41431-019-0540-0 (englisch).
- L. M. Henricks, C. A. T. C. Lunenburg, F. M. de Man, D. Meulendijks, G. W. J. Frederix, E. Kienhuis, G. J. Creemers, A. Baars, V. O. Dezentjé, A. L. T. Imholz, F. J. F. Jeurissen, J. E. A. Portielje, R. L. H. Jansen, P. Hamberg, A. J. Ten Tije, H. J. Droogendijk, M. Koopman, P. Nieboer, M. H. W van de Poel, C. M. P. W. Mandigers, H. Rosing, J. H. Beijnen, E. V. Werkhoven, A. B. P. van Kuilenburg, R. H. N. van Schaik, R. H. J. Mathijssen, J. J. Swen, H. Gelderblom, A. Cats, H. J. Guchelaar, J. H. M. Schellens: DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. In: Lancet Oncol. Band 19, Nr. 11, November 2018, S. 1459–1467, doi:10.1016/S1470-2045(18)30686-7 (englisch).
- Europäische Arzneimittel-Agentur: EMA recommendations on DPD testing prior to treatment with fluorouracil, capecitabine, tegafur and flucytosine. (PDF) 2020, abgerufen am 28. Dezember 2020.
- Diana Lüftner, Sara Brucker, Bernhard Wörmann: Test auf DPD-Mangel vor dem Einsatz von Fluoropyrimidinen wie Capecitabine als Kassenleistung. In: Senologie - Zeitschrift für Mammadiagnostik und -therapie. Band 17, Nr. 04, Dezember 2020, ISSN 1611-6453, S. 247–248, doi:10.1055/a-1290-2074.
- L. M. Henricks, F. L. Opdam, J. H. Beijnen, A. Cats, J. H. M. Schellens: DPYD genotype-guided dose individualization to improve patient safety of fluoropyrimidine therapy: call for a drug label update. In: Annals of Oncology. Band 28, Nr. 12, Dezember 2017, S. 2915–2922, doi:10.1093/annonc/mdx411 (elsevier.com [abgerufen am 28. Dezember 2020]).