Wirkstoffdesign
Wirkstoffdesign (auch Rationales Wirkstoffdesign) bezeichnet den gezielten Entwurf von Wirkstoffen. Diese Wirkstoffe dienen unter anderem der Entwicklung von Arzneimitteln oder Pflanzenschutzmitteln. Der gezielte Entwurf von Wirkstoffen basiert auf dem Auffinden und Optimieren von Leitstrukturen, die als Teil eines Liganden an ein Target binden.[1] Wirkstoffdesign ist eine Form des rationalen Designs.
Eigenschaften
Meistens ist ein Wirkstoff ein small molecule, ein rekombinantes Protein oder ein Vektor, das eine Wirkung in Lebewesen hervorruft. Diese Wirkung erfolgt meistens durch Bindung des Wirkstoffs an ein Protein, wodurch dessen Wirkung verstärkt oder geschwächt werden kann. Die Wirkstoffe sind oftmals in Form und Ladung komplementär zu der Bindungsstelle am Protein.[2] (Man beschreibt das Ganze mit dem Schlüssel-Schloss-Prinzip.) Wirkstoffe mit ähnlicher Form und Ladung (Strukturanaloga) besitzen meistens ähnliche Wirkungen. Das Wirkstoffdesign wird oftmals durch molekulare Modellierung wie eine PK/PD-Modellierung unterstützt.[3] Im Gegensatz zum klassischen Hochdurchsatz-Screening von Molekül-Bibliotheken verwendet das Wirkstoffdesign Erkenntnisse über die Molekülstrukturen von Ligand und target. Für ein Wirkstoffdesign muss die Aktivierung oder Hemmung eines targets mit einer Wirkung korrelieren und das target auch die Bindung eines Liganden von der Form her ermöglichen (engl. druggability ‚Wirkstoff-Bindungsfähigkeit‘).[4] Im engeren Sinn bezeichnet Wirkstoffdesign den Entwurf von Liganden für ein Target, das in Folge der Bindung des Pharmakophors erst eine Wirkung hervorruft.[5] Das Wirkstoffdesign umfasst die Anpassung von Eigenschaften des Wirkstoffes wie Pharmakodynamik, effektive Dosis, (idealerweise orale) Bioverfügbarkeit, Plasmahalbwertszeit, Verstoffwechselung, protektiver Quotient, Toxizität und unerwünschte Arzneimittelwirkungen. Hierfür existieren verschiedene Methoden wie die Rule of Five, die lipophilic efficiency oder SPORCalc.[6]
Typen
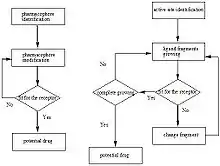
Das Wirkstoffdesign besitzt zwei verschiedene, zum Teil auch kombinierbare Herangehensweisen, das Liganden-basierte und das Struktur-basierte Wirkstoffdesign.
Liganden-basiert
Ligand-basiertes Wirkstoffdesign (auch indirektes Wirkstoffdesign) verwendet Wirkstoffe, die Wirkstoffen in Form und Ladung ähneln, bei denen eine Wirkung empirisch bekannt ist. Die Wirkstoffe werden durch Hochdurchsatz-Screening identifiziert. Aus deren Molekülstruktur kann auf die Form der Bindungsstelle am target zurückgeschlossen und eine Quantitative Struktur-Wirkungs-Beziehung ermittelt werden.[7] Dies ermöglicht eine begrenzte Vorhersage der Bindungen anderer Stoffe an diese Stelle.
Struktur-basiert
Das Struktur-basierte Wirkstoffdesign (auch direktes Wirkstoffdesign) basiert auf der Kenntnis der Molekülstruktur und der des targets (Proteinstruktur), die durch Kristallstrukturanalyse oder Kernspinresonanzspektroskopie gewonnen wurden.[8]
Innerhalb des Struktur-basierten Ansatzes kann ein Wirkstoff über eine Datenbanksuche identifiziert werden (sofern darin vorhanden) oder aufgrund der Kenntnis der Struktur des targets konstruiert und anschließend synthetisiert werden. Dies reduziert die durchschnittliche Anzahl an Versuchsansätzen.[9][10][11] Beim Scaffold Hopping wird das nichtbindende Gerüst des Pharmakophors variiert.
Bestimmung der Bindungsstelle
Durch Proteincharakterisierung wird die Bindungsstelle für das Pharmakophor identifiziert, z. B. ein aktives Zentrum eines Enzyms. Dabei werden die an der Bindung teilnehmenden Atome untersucht, wofür meistens die Struktur des targets bei gebundenem Liganden bestimmt wird. Die typischen Wechselwirkungen werden durch typische Atome vermittelt:
- hydrophobe Atome in aliphatischen und aromatischen Aminosäuren
- Donor einer Wasserstoffbrückenbindung durch ein Sauerstoff- oder Stickstoffatom mit Wasserstoff
- Akzeptor einer Wasserstoffbrückenbindung durch ein Sauerstoff- oder Stickstoffatom mit freiem Elektronenpaar
- Polare Atome ohne Wasserstoffbeteiligung wie Sauerstoff, Stickstoff, Schwefel, Phosphor, Chlor, Iod, Metallionen und polarisierte Kohlenstoffatome (mit vorhergehenden Heteroatomen verbunden).
Ligandenfragmente

Die Struktur des Liganden kann auch im Zuge einer Modellierung in die Formen seiner Bestandteile (Formmotive) unterteilt werden, die auch als Fragmente bezeichnet werden.[12] Durch aufeinanderfolgendes Hinzufügen auf das erste Fragment (engl. seed ‚Keimling‘) wächst die Größe des so entworfenen Liganden. Zwar gibt es nur wenige Fragmente, jedoch können durch deren Kombination viele verschiedene Moleküle erzeugt werden. Zur Ermittlung der potenziellen Energiefläche werden mit Großrechnern die freie Energie der jeweiligen Bindung berechnet, um die niedrigste potentielle Energie zwischen der Bindungsfläche und dem Pharmakophor zu identifizieren. Zur Einsparung von Rechnerzeit und -kapazitäten werden die Schritte priorisiert, z. B. werden fest bindende Fragmente bei der Berechnung bevorzugt. Nach Ermittlung bindenender Fragmente für mehrere Bindungsstellen werden diese zu einem Molekül vereinigt. Die Konformation niedrigster potenzieller Energie ermöglicht eine höher affine Bindung des Liganden.[9][10][13]
Bewertungsmethoden
Das Struktur-basierte Wirkstoffdesign berechnet die optimalen Liganden vor allem in Bezug auf eine möglichst affine Bindung an das target. Eine Bewertungsmethode betrachtet die mit der Anlagerung verbundene Freie Enthalpie:[14]

die freie Enthalpie setzt sich dabei aus vier Elementen zusammen:

- Desolvatation – Enthalpie der Entfernung des Liganden aus dem Lösungsmittel
- Molekularbewegung – Herabsetzung der Entropie durch Minderung der Freiheitsgrade bei der Bindung
- Konfiguration – nötige Konformationsänderungen zur Bindung
- Interaktion – Enthalpiegewinn durch die Wechselwirkungen an der Kontaktfläche
Für jede der freien Enthalpien bzw. Energien können unterschiedliche Berechnungsmethoden verwendet werden.[15][16][17]
Weblinks
Einzelnachweise
- Madsen, Ulf; Krogsgaard-Larsen, Povl; Liljefors, Tommy: Textbook of Drug Design and Discovery. Taylor & Francis, Washington, DC 2002, ISBN 0-415-28288-8.
- X. Zheng, L. Gan, E. Wang, J. Wang: Pocket-based drug design: exploring pocket space. In: AAPS J. (2013), Band 15, Nr. 1, S. 228–241. doi:10.1208/s12248-012-9426-6. PMID 23180158; PMC 3535113 (freier Volltext).
- Cohen, N. Claude: Guidebook on Molecular Modeling in Drug Design. Academic Press, Boston 1996, ISBN 0-12-178245-X.
- Y. Yuan, J. Pei, L. Lai: Binding site detection and druggability prediction of protein targets for structure-based drug design. In: Curr Pharm Des. (2013), Band 19, Nr. 12, S. 2326–2333. PMID 23082974.
- Tollenaere JP: The role of structure-based ligand design and molecular modelling in drug discovery. In: Pharm World Sci. 18, Nr. 2, April 1996, S. 56–62. doi:10.1007/BF00579706. PMID 8739258.
- Smith J, Stein V: SPORCalc: A development of a database analysis that provides putative metabolic enzyme reactions for ligand-based drug design. In: Computational Biology and Chemistry. 33, Nr. 2, April 2009, S. 149–59. doi:10.1016/j.compbiolchem.2008.11.002. PMID 19157988.
- Guner, Osman F.: Pharmacophore Perception, Development, and use in Drug Design. International University Line, La Jolla, Calif 2000, ISBN 0-9636817-6-1.
- Leach, Andrew R.; Harren Jhoti: Structure-based Drug Discovery. Springer, Berlin 2007, ISBN 1-4020-4406-2.
- Wang R,Gao Y,Lai L: LigBuilder: A Multi-Purpose Program for Structure-Based Drug Design. In: Journal of Molecular Modeling. 6, Nr. 7–8, 2000, S. 498–516. doi:10.1007/s0089400060498.
- Schneider G, Fechner U: Computer-based de novo design of drug-like molecules. In: Nat Rev Drug Discov. 4, Nr. 8, August 2005, S. 649–63. doi:10.1038/nrd1799. PMID 16056391.
- Jorgensen WL: The many roles of computation in drug discovery. In: Science. 303, Nr. 5665, März 2004, S. 1813–8. bibcode:2004Sci...303.1813J. doi:10.1126/science.1096361. PMID 15031495.
- A. Kumar, A. Voet, K. Y. Zhang: Fragment based drug design: from experimental to computational approaches. Curr Med Chem. (2012), Band 19, Nr. 30, S. 5128–5147. PMID 22934764.
- Verlinde CL, Hol WG: Structure-based drug design: progress, results and challenges. In: Structure. 2, Nr. 7, Juli 1994, S. 577–87. doi:10.1016/S0969-2126(00)00060-5. PMID 7922037.
- Böhm HJ: The development of a simple empirical scoring function to estimate the binding constant for a protein-ligand complex of known three-dimensional structure. In: J. Comput. Aided Mol. Des.. 8, Nr. 3, Juni 1994, S. 243–56. bibcode:1994JCAMD...8..243B. doi:10.1007/BF00126743. PMID 7964925.
- Gohlke H, Hendlich M, Klebe G: Knowledge-based scoring function to predict protein-ligand interactions. In: J. Mol. Biol.. 295, Nr. 2, Januar 2000, S. 337–56. doi:10.1006/jmbi.1999.3371. PMID 10623530.
- Clark RD, Strizhev A, Leonard JM, Blake JF, Matthew JB: Consensus scoring for ligand/protein interactions. In: J. Mol. Graph. Model.. 20, Nr. 4, Januar 2002, S. 281–95. doi:10.1016/S1093-3263(01)00125-5. PMID 11858637.
- Wang R, Lai L, Wang S: Further development and validation of empirical scoring functions for structure-based binding affinity prediction. In: J. Comput. Aided Mol. Des.. 16, Nr. 1, Januar 2002, S. 11–26. bibcode:2002JCAMD..16...11W. doi:10.1023/A:1016357811882. PMID 12197663.