Multisystematrophie
Der Begriff Multisystematrophie (MSA) bezeichnet eine rasch fortschreitende neurodegenerative Erkrankung, bei der multiple Systeme betroffen sind. Der Begriff wurde 1969 von Graham und Oppenheimer geprägt, die erkannten, dass olivopontocerebelläre Atrophie (OPCA) (Dejerine-Thomas-Syndrom), striatonigrale Degeneration (SND) und die idiopathische orthostatische Hypotonie (Shy-Drager-Syndrom) Ausprägungen derselben Erkrankung sind. Papp, Kahn und Lantos beschrieben 1989 Oligodendrogliale Einschlusskörper (Glial Cytoplasmic Inclusions, GCI) als gemeinsamen histologischen Marker.[1] Mit der Entdeckung von α-Synuclein als Hauptkomponente der GCI wird die MSA wie der Morbus Parkinson und die Lewy-Körperchen-Demenz zu der Gruppe der Synucleinopathien gezählt.
| Klassifikation nach ICD-10 | |
|---|---|
| G90.3 | Multisystem-Atrophie, |
| G23.1 | Progressive supranukleäre Ophthalmoplegie [Steele-Richardson-Olszewski-Syndrom] |
| G23.2 | Multiple Systematrophie vom Parkinson-Typ [MSA-P] |
| G23.3 | Multiple Systematrophie vom zerebellären Typ [MSA-C] |
| G23.8 | Sonstige näher bezeichnete degenerative Krankheiten der Basalganglien: Kalzifikation der Basalganglien: Neurogene orthostatische Hypotonie [Shy-Drager-Syndrom] |
| ICD-10 online (WHO-Version 2019) | |
Epidemiologie
Die Prävalenz beträgt etwa 4,4 Fälle auf 100.000 Einwohner.[2] Damit kommen auf 100 Fälle mit Morbus Parkinson etwa 2,2 Fälle mit MSA. Die Geschlechterverhältnisse sind in etwa ausgeglichen. Während in Europa die MSA-P die häufigere Variante ist (49 %[3]), überwiegt in Japan die MSA-C.
MSA tritt in der Regel zwischen dem 40. und 60. Lebensjahr auf, der Erkrankungsgipfel liegt bei 57 Jahren.[4] Die Erkrankung ist rasch fortschreitend und führt innerhalb von 3–5 Jahren zum Verlust der Gehfähigkeit und im Mittel nach 8–10 Jahren zum Tode.[5]
Symptome
Klinische Symptome sind vor allem extrapyramidal-motorische Symptome im Sinne eines Parkinsonismus mit Bradykinesie, Tremor und Rigor, zerebelläre Symptome wie z. B. Dysmetrie, Nystagmus und Gang- und Stand-Ataxie mit Fallneigung, aber auch Schluckstörungen (Dysphagie) und Sprechstörungen (Dysarthrie), sowie Regulationsstörungen des autonomen Nervensystems wie Orthostatische Hypotonie, Harninkontinenz und erektile Dysfunktion. Zusätzlich finden sich häufig eine subkortikale Demenz,[6] Pyramidenbahnzeichen wie Hyperreflexie oder positiver Babinski-Reflex. Je nachdem, welche Systeme betroffen sind, tritt nur ein Teil der beschriebenen Symptome auf. Nach den gültigen Diagnosekriterien von 1998 werden zwei motorische Phänotypen unterschieden: Überwiegen die Parkinson-Symptome, spricht man von MSA-P, überwiegen zerebelläre Symptome, spricht man von MSA-C. Eine autonome Dysfunktion ist Voraussetzung für die Diagnose einer MSA.[7]
Störungen der Schluckfunktionen führen nicht selten zu Aspirationspneumonien, was auch zur verkürzten Lebenserwartung beiträgt.
Diagnostik
Die Diagnose beruht hauptsächlich auf Anamnese und klinischer Untersuchung, die Differentialdiagnose kann aber auch für Experten schwierig sein. Neben einer obligaten autonomen Dysfunktion ist fehlendes Ansprechen auf L-DOPA ein wichtiges Kriterium zur Abgrenzung vom Morbus Parkinson. Einige klinische Zeichen sind als Hinweis auf eine mögliche Multisystematrophie zu beachten, wie Dysphagie, Dysarthrie, inspiratorischer Stridor, frühe Fallneigung und schneller Krankheitsfortschritt.[8]
In Computertomographie-Aufnahmen kann in einigen Fällen eine Atrophie des Kleinhirnes oder der Brücke (Pons) beobachtet werden. In Kernspinaufnahmen sieht man gelegentlich eine Atrophie des Putamens. Wichtig sind diese Untersuchungen in erster Linie jedoch zur Abgrenzung gegenüber anderen neurologischen Krankheitsbildern.
Besonders wichtig kann in schwierigen Fällen das IBZM-SPECT sein, mit dem die MSA meistens gegenüber dem Morbus Parkinson abgegrenzt werden kann. Mit diesem wird das Fehlen postsynaptischer Dopaminrezeptoren nachgewiesen. Beim Morbus Parkinson dagegen sind die postsynaptischen Rezeptoren häufig normal, hier findet sich ein Defekt der dopaminergen Neurone.
Pathologie
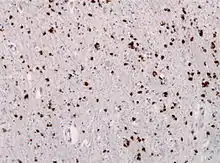
Im Gehirn betroffener Patienten finden sich eine abnorme Ansammlung des Proteins α-Synuclein in Einschlusskörpern in der Oligodendroglia (auch als Papp-Lantos-Einschlüsse bekannt), daher wird die MSA wie der Morbus Parkinson und die Lewy-Körperchen-Demenz zu der Gruppe der Synucleinopathien gezählt. Zusätzlich beobachtet man einen Untergang von Nervenzellen mit reaktiver Gliose.
Die Degeneration der Substantia nigra führt zu Parkinsonismus, diejenige des Striatums zum Verlust von Dopamin-Rezeptoren und folglich zum Verlust des Ansprechens auf dopaminerge Therapie.
Die Ataxie wird verursacht durch eine Atrophie des Kleinhirnes, häufig zusammen mit einer Atrophie von Oliven (Nucleus olivaris) und der Brücke.
Ein Untergang von präsynaptischen sympathischen Neuronen im Rückenmark wird für die autonome Dysfunktion verantwortlich gemacht; es gibt aber auch Hinweise auf eine begleitende postsynaptische Lewy-Körperchen-Pathologie.[9]
Therapie
Die Therapie der Multisystematrophie ist wie die Erkrankung eine multisystemische. Für die zerebelläre Symptomatik existiert derzeit keine Therapie.
Dopaminergika. Parkinsonismus wird mit Dopaminergika behandelt, wobei L-DOPA etwas besser wirksam ist als Dopaminagonisten. Zur Unterstützung können MAO-Hemmer (Monoaminooxidase-Hemmer) und COMT-Hemmer (Catechol-O-Methyltransferase-Hemmer) eingesetzt werden, die den Dopaminabbau hemmen und somit die Wirksamkeit verlängern. Prinzipiell zeigen aber nur ein Drittel der MSA-Patienten ein Ansprechen auf dopaminerge Therapie; dieses hält im Mittel drei Jahre an. Amantadin, ein NMDA-Rezeptor-Antagonist, wird aufgrund seiner Wirksamkeit gegen Dyskinesien und einer möglichen Antiparkinson-Wirkung häufig eingesetzt.
Autonome Dysfunktion. Die Therapie der autonomen Dysfunktion ist sowohl nicht-medikamentös als auch medikamentös. Die orthostatische Hypotension lässt sich mit Stützstrümpfen, erhöhter Salz- und Wasserzufuhr und Schlafen mit erhöhtem Kopfkissen bessern; medikamentös stehen Fludrocortison und Midodrin zur Auswahl. Harninkontinenz kann in leichten Fällen mit Einlagen behandelt werden; medikamentös stehen Trospiumchlorid, Oxybutynin und Tolterodin zur Verfügung. Liegt eine inkomplette Blasenentleerung vor, muss katheterisiert werden. Zur Therapie der erektilen Dysfunktion eignen sich Sildenafil oder ähnliche PDE-5-Hemmer.[10]
Palliation. Palliative Maßnahmen können bei schweren Schluckstörungen notwendig werden wie künstliche Ernährung mittels PEG-Sonde (Perkutane endoskopische Gastrostomie). Bei starkem Stridor kann eine CPAP-Beatmung Besserung bringen.
Depression. Die häufig begleitende Depression sollte ebenfalls mitbehandelt werden.
Immunglobuline. Die beteiligten toxischen Zytokine lassen vermuten, dass Inflammation eine Rolle in der Pathogenese spielt. Deswegen gibt es zumindest teilweise erfolgreiche Versuche, die Erkrankung mit einer intravenösen Immunglobulin-Therapie zu behandeln.[11]
Einzelnachweise
- M. I. Papp, J. E. Kahn, P. L. Lantos: Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). In: J Neurol Sci. 94(1-3), Dez 1989, S. 79–100. PMID 2559165
- A. Bjornsdottir, G. Gudmundsson, H. Blondal, E. Olafsson: Incidence and prevalence of multiple system atrophy: a nationwide study in Iceland. In: J Neurol Neurosurg Psychiatry. 28. Nov 2012.
- F. Geser u. a.: The European Multiple System Atrophy-Study Group (EMSA-SG). In: J Neural Transm. 112(12), Dez 2005, S. 1677–1686. Epub 2005 Jul 29.
- G. K. Wenning, C. Colosimo, F. Geser, W. Poewe: Multiple system atrophy. In: The Lancet Neurology. 3(2), Feb 2004, S. 93–103. Review. PMID 14747001
- H. Watanabe u. a.: Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. In: Brain. 125(Pt 5), Mai 2002, S. 1070–1083. Review. PMID 11960896
- T. H. Bak, L. M. Crawford, V. C. Hearn, P. S. Mathuranath, J. R. Hodges: Subcortical dementia revisited: similarities and differences in cognitive function between progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and multiple system atrophy (MSA). In: Neurocase. 11(4), 2005, S. 268–273.
- S. Gilman u. a.: Consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and American Academy of Neurology. In: Clin Auton Res. 8(6), Dez 1998, S. 359–362. Review. PMID 9869555
- M. Köllensperger u. a.: Red flags for multiple system atrophy. In: Mov Disord. 23(8), 15. Jun 2008, S. 1093–1099. PMID 18442131
- S. Orimo, T. Kanazawa, A. Nakamura, T. Uchihara, F. Mori, A. Kakita, K. Wakabayashi, H. Takahashi: Degeneration of cardiac sympathetic nerve can occur in multiple system atrophy. In: Acta Neuropathol. 113(1), Jan 2007, S. 81–86. Epub 2006 Nov 7. PMID 17089131
- I. F. Hussain, C. M. Brady, M. J. Swinn, C. J. Mathias, C. J. Fowler: Treatment of erectile dysfunction with sildenafil citrate (Viagra) in parkinsonism due to Parkinson's disease or multiple system atrophy with observations on orthostatic hypotension. In: J Neurol Neurosurg Psychiatry. 71(3), Sep 2001, S. 371–374. PMID 11511713.
- P. Novak, A. Williams, P. Ravin, O. Zurkiya, A. Abduljalil, V. Novak: Treatment of multiple system atrophy using intravenous immunoglobulin. In: BMC Neurol. 12(1), 1. Nov 2012, S. 131.
Literatur
- U. Wüllner, T. Klockgether: Klinik und Therapie der Multisystematrophie. In: Dtsch Arztebl. 100(7), 2003, S. A-408 / B-357 / C-339.