Morbus Castleman
Morbus Castleman (Castleman-Krankheit) ist eine heterogene Gruppe von Tumoren der Lymphknoten, die auch als angiofollikuläre Lymphknoten-Hyperplasie[1] oder Riesenlymphknoten-Hyperplasie bezeichnet werden.
| Klassifikation nach ICD-10 | |
|---|---|
| D47.7 | Sonstige näher bezeichnete Neubildungen unsicheren oder unbekannten Verhaltens des lymphatischen, blutbildenden und verwandten Gewebes |
| ICD-10 online (WHO-Version 2019) | |
Einteilung
Auch wenn alle Tumoren ähnliche histologische Eigenschaften teilen, gibt es doch unterschiedliche Formen mit unterschiedlicher Symptomatik, Therapie und Prognose. Klinisch lassen sich verschiedene Formen des M. Castleman unterscheiden. Neben der lokalisierten („unizentrischen“) Form mit lediglich einem vergrößerten Lymphknoten gibt es multizentrische Formen, bei der Lymphknoten in mehreren Regionen betroffen sind.[2]
Derzeit ist folgende Klassifikation gebräuchlich:[3]
- Unizentrische Formen (UCD)
- HHV-8 (Humanes Herpesvirus 8) assoziierte multizentrische Form
- Idiopathische multizentrische Form (iMCD)
Unizentrische Formen (UCD)
Der erste Fall einer solchen peculiar form of lymph-node hyperplasia wurde 1954 durch den US-amerikanischen Pathologen Benjamin Castleman auf einer klinisch-patholgische Fallkonferenz im Massachusetts General Hospital vorgestellt.,[4] Die weitere Beschreibung der unizentrischen Form erfolgte 1956 durch Castleman[5] der 1972 dann zwei histologische Formen unterschied:
- hyalin-vaskulärer Typ in 91 %: meist besteht ein mediastinaler Tumor ohne weitere systemische Symptome. Dieser Typ gilt als gutartiger klonaler Tumor der follikulären dendritischen Zellen. Es können auch Zell-Dysplasien auftreten, auch cytogenetische Veränderungen wurden gelegentlich beschrieben. Selten kann die hyalin-vaskuläre Form zu einem Dendritenzell-Sarkom entarten.
- Plasmazell-Typ in 9 %, der häufiger außerhalb des Mediastinums auftritt und öfter systemische Symptome aufweist, wie Anämie oder Hypergammaglobulinämie.
Multizentrische Formen (MCD)
Bei dem multizentrischen Plasmazell-Typ kann eine idiopathische Form (iMCD) von einer Form abgegrenzt werden, die mit einer Infektion durch das Humane Herpesvirus 8 (HHV-8) assoziiert ist (HHV-8-assoziierte MCD). Diese Form tritt besonders bei HIV-positiven Patienten auf und gilt als lymphoproliferative Erkrankung mit Gefahr einer Progression zu einem Lymphom. Dabei infizieren die HHV-8-Viren B-Zellen und diese proliferieren als große Plasmablasten.
Die multizentrische Form besteht zumeist aus dem Plasmazell-Typ. Varianten mit Eigenschaften des hyalin-vaskulären Typs kommen vor, diese sind zumeist mit dem TAFRO-Syndrom assoziiert und wird als hypervaskuläre Variante bezeichnet. Das TAFRO-Syndrom bestehend aus Thrombozytopenie, Anasarka, Fieber, retikuläre Fibrose und Organomegalie wird als Variante der iMCD angesehen.[6] Darüber hinaus gibt es einen gemischten Typ.[7]
Bei MCD kommt es unter anderem zu einer vermehrten Ausschüttung des proinflammatorischen Zytokins Interleukin-6. Bei der HHV-8-assoziierten Form geschieht dies durch virale Produkte, bei der idiopathischen Form ist die Ursache der Interleukin-6-Ausschüttung nicht geklärt. Interleukin-6 ist verantwortlich für die meisten systemischen Symptome. Interleukin-6 stimuliert auch die B-Zell-Proliferation, wirkt angiogenetisch und induziert eine Akute-Phase-Reaktion.
Epidemiologie
Die Krankheit tritt unabhängig von Alter und Geschlecht auf. Es wird geschätzt, dass weniger als 1:100.000 von der Krankheit betroffen ist, wobei die lokalisierte Form deutlich häufiger auftritt. Am häufigsten sind das Mediastinum, aber auch der Bauchraum, besonders der Retroperitonealraum, und die oberflächlichen Lymphknoten betroffen.[1] Die einzelne Läsion kann auf mehrere Zentimeter Durchmesser anwachsen.
Symptome
Die lokalisierte Form verursacht bei ungefähr der Hälfte der Patienten keine systemischen Beschwerden. Der Tumor wird oft zufällig ("inzidentiell") entdeckt oder verursacht lokale Verdrängungssymptome durch Druck auf Gefäße oder Nerven und hierdurch Thorax- oder Bauchschmerzen. Ansonsten kann eine "B-Symptomatik" mit Abgeschlagenheit, Fieber, Gewichtsverlust auftreten.[1]
Multizentrische Verlaufsformen verursachen meist Symptome, die durch die vermehrte Interleukin-6-Ausschüttung verursacht werden: Gewichtsverlust (69 %), Fieber (67 %), periphere Lymphadenopathie (81 %), Hepato- und/oder Splenomegalie (74 %), Müdigkeit, Hypoalbuminämie und Hypergammaglobulinämie.
Bei mehr als 60 % aller von HIV betroffenen M.-Castleman-Patienten konnte das HHV-8 nachgewiesen werden, während hingegen HIV-negative Castleman-Patienten nur in 20 bis 40 % der Fälle HHV-8-positiv sind.[1]
Gelegentlich ist der Morbus Castleman mit anderen Erkrankungen assoziiert, auf die besonders geachtet werden sollte:
- Kaposi-Sarkom, eine ebenfalls durch das Humane Herpesvirus 8 ausgelöste Hautkrebsform bei der HHV-8-positiven Form des Plasmazell-Typs
- POEMS-Syndrom bestehend aus Polyneuropathie, Organomegalie, Endokrinopathie, M-Gradient in der Serumelektrophorese und Hautveränderungen (skin changes), in bis zu 23 %, beim Plasmazell-Typ
- osteosklerotisches Plasmozytom beim Plasmazell-Typ
- Paraneoplastischer Pemphigus beim hyalin-vaskulären Typ
- Sarkom der follikulär dendritischen Zellen beim hyalin-vaskulären Typ
Diagnose
Die Diagnose erfolgt durch eine Biopsie mit nachfolgender histologischer und immunhistochemischer Untersuchung. Diese kann bei unizentrischen Formen als kurative Exzisionsbiopsie erfolgen.
Histologie
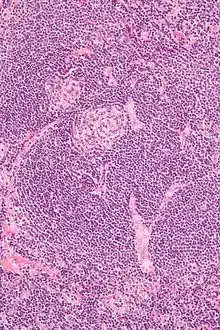
Histologisch besteht der vergrößerte Lymphknoten aus einer deutlich vermehrten Anzahl von Lymphfollikeln, die Sinus sind durch die Expansion der Follikel vermindert oder nicht mehr erkennbar, und die Kapsel ist verdickt. In den Follikeln sind die Keimzentren atroph oder regressiv verändert. Es finden sich aber oft mehrere Keimzentren in einer Follikel-Mantelzone, sog. twinning. Gelegentlich kann man senkrecht in den Follikel eintretende hyalinisierte Gefäße erkennen, sog. Lollipop-Phänomen. Die Mantelzone ist verdickt und hat ein zwiebelschalenartiges Aussehen (onion-skinning). Dies entsteht durch eine Ansammlung von Mantelzonen-Lymphozyten entlang der follikulären dendritischen Zellen rund um die atrophen Keimzentren. In der interfollikulären Zone finden sich vermehrt endotheliale Venolen sowie kleine Lymphozyten und gelegentlich Plasmazellen.
Während alle beschriebenen histologischen Veränderungen einzeln auch bei anderen reaktiven und neoplastischen Lymphknotenveränderungen (wie Lymphomen) vorkommen können, ist ihr Zusammentreffen typisch und diagnostisch für den Morbus Castleman (ICD-10: D36.0)
Behandlung
Unizentrische Formen
Im Jahr 2020 wurde die erste internationale Leitlinie zur Behandlung der Castleman-Erkrankung publiziert.[8] Darauf gründen sich die folgenden Empfehlungen:
Bei der lokalisierten unizentrischen Form besteht die Therapie in der vollständigen chirurgischen Entfernung. Dies war in einer Übersichtsarbeit mit 278 Fällen bei 90 % möglich und zeigte eine krankheitsfreie Fünfjahres-Überlebensrate von über 80 % mit einer Gesamtüberlebensrate von über 90 %. Weniger als 5 % der Patienten verstarb in den ersten zehn Jahren an einem unizentrischen Morbus Castleman.[9]
Sofern systemischen Symptome vorhanden waren, bilden sich diese in der Regel nach der vollständigen chirurgischen Resektion zurück. In einigen Fällen (zwischen 3,8 und 16,2 % der Fälle[8]) bleiben die Symptome jedoch bestehen. Hier muss zunächst gründlich geprüft werden, ob eine andere Erkrankung bisher übersehen wurde, z. B. iMCD, Lymphome, Autoimmunerkrankungen. Sollte keine alternative Ursache gefunden werden, kann eine weiterführende systemische Behandlung erforderlich sein.
Ist die unizentrische Lymphknotenhyperplasie chirurgisch nicht resezierbar, ist abzuwägen, wie weiter behandelt wird. Bei asymptomatischen Patienten ohne Risikofaktoren kann ein rein abwartendes Verhalten mit regelmäßigen Kontrolluntersuchungen indiziert sein.
Andernfalls kann eine systemische Therapie oder eine Embolisierung geeignet sein, den Tumor resektierbar zu machen. Bei Symptomen die überwiegend aus Druck auf umliegende Strukturen resultieren, erscheint dazu Rituximab ggf. in Verbindung mit Glucocorticoiden geeignet. Bei systemischen Symptomen bei erhöhter Blutsenkungsgeschwindigkeit, IL6, CRP oder Anämie sollen zudem Siltuximab oder Tocilizumab vor schwerwiegenderen Eingriffen versucht werden.
Für den Einsatz von Radiotherapie gibt es wegen der Langzeitfolgen keinen Konsens. Zunächst sollten die zuvor genannten Möglichkeiten ausgeschöpft sein. Selbst dann ist gegenüber anderen Behandlungen wie Sirolimus, Glucocorticoiden und Cyclosporin A abzuwägen.[8]
Multizentrische Formen
Bei der multizentrischen Form erfolgt eine systemische Therapie. Da die Interleukin-6-Ausschüttung der Pathogenese und den Symptomen zugrunde liegt, wurde der gegen Interleukin-6 gerichtete monoklonale Antikörper Siltuximab erfolgreich in einer randomisierten kontrollierten Studie eingesetzt[10] und 2014 von der US-amerikanischen Zulassungsbehörde Food and Drug Administration (FDA) und der Europäischen Arzneimittel-Agentur EMA[11] zur Behandlung der HHV-8-negativen multizentrischen Form zur Standardtherapie zugelassen.[12]
Bei multizentrischem Morbus Castleman mit Nachweis einer HHV-8-Infektion erfolgt die Produktion des Interleukins-6 durch die HHV-8-Viren selbst, weshalb der Einsatz des monoklonalen Antikörpers Siltuximab nicht sinnvoll ist. Neuere Studien wurden mit dem gegen das B-Lymphozytenantigen CD20 gerichteten monoklonalen Antikörper Rituximab durchgeführt und zeigten eine erheblich höhere Remissionsrate mit länger andauernder Remission und einer relativ geringen Rate unerwünschter Wirkungen. Rituximab ist infolgedessen die Standardtherapie. Erst bei fehlendem Ansprechen auf die Therapie oder einem sehr schweren Verlauf werden Chemotherapien, ggf. in Verbindung mit Rituximab versucht.[13]
Eine eventuell zugrundeliegende HIV-Erkrankung wird mit antiretroviralen Medikamenten behandelt. Die Prognose des multizentrischen HHV-8-positiven M. Castleman ist schlecht. Sie betrug 1996 in einer Studie bei HIV-infizierten Patienten im Mittel nur 14 Monate.[14] Dies hat sich durch die neuen Therapiemöglichkeiten jedoch vermutlich erheblich verbessert.[13]
Literatur
- U.-N. Riede: Allgemeine und spezielle Pathologie. Georg Thieme, 2004, ISBN 3-13-683305-8, S. 553ff.
- Michael L. Blute, Jeremy S. Abramson, Kevin C. Cronin, Valentina Nardi: Case 5-2017: A 19-year-old man with hematuria and a retroperitoneal mass. New England Journal of Medicine 2017, Band 376, Ausgabe 7 vom 16. Februar 2017, S. 684–692, doi:10.1056/NEJMcpc1610100 – Fallbeschreibung eines unizentrischen hyalin-vaskulären Castleman-Tumors im Retroperitonealraum.
Einzelnachweise
- Morbus Castleman. In: Orphanet (Datenbank für seltene Krankheiten).
- D. C. Fajgenbaum, D. Shilling: Castleman Disease Pathogenesis. In: Hematology/oncology clinics of North America. Band 32, Nummer 1, 02 2018, S. 11–21, doi:10.1016/j.hoc.2017.09.002, PMID 29157613 (Review).
- E. Oksenhendler, D. Boutboul, D. Fajgenbaum, A. Mirouse, C. Fieschi, M. Malphettes, L. Vercellino, V. Meignin, L. Gérard, L. Galicier: The full spectrum of Castleman disease: 273 patients studied over 20 years. In: British Journal of Haematology. Band 180, Nummer 2, 01 2018, S. 206–216, doi:10.1111/bjh.15019, PMID 29143319.
- B. Castleman, V. W. Towne: Case records of the Massachusetts General Hospital: Case No. 40231. In: N Engl J Med. 250(23), 10. Jun 1954, S. 1001–1005. PMID 13165944
- B. Castleman, L. Iverson, V. P. Menendez: Localized mediastinal lymphnode hyperplasia resembling thymoma. In: Cancer. Band 9, 1956, S. 822–830.
- J. M. Hawkins, V. Pillai: TAFRO syndrome or Castleman-Kojima syndrome: a variant of multicentric Castleman disease. In: Blood. 126, 2015, S. 2163, doi:10.1182/blood-2015-07-662122
- David C. Fajgenbaum, Thomas S. Uldrick, Adam Bagg, Dale Frank, David Wu: International, evidence-based consensus diagnostic criteria for HHV-8–negative/idiopathic multicentric Castleman disease. In: Blood. Band 129, Nr. 12, 23. März 2017, ISSN 0006-4971, S. 1646–1657, doi:10.1182/blood-2016-10-746933.
- Frits van Rhee, Eric Oksenhendler, Gordan Srkalovic, Peter Voorhees, Megan Lim: International evidence-based consensus diagnostic and treatment guidelines for unicentric Castleman disease. In: Blood Advances. Band 4, Nr. 23, 7. Dezember 2020, ISSN 2473-9529, S. 6039–6050, doi:10.1182/bloodadvances.2020003334.
- N. Talat, A. P. Belgaumkar, K. M. Schulte: Surgery in Castleman's disease: a systematic Review of 404 published cases. In: Annals of Surgery. Band 255, 2012, S. 677–684.
- F. van Rhee, R. S. Wong, N. Munshi u. a.: Siltuximab for multicentric Castleman's disease: a randomised, double-blind, placebo-controlled trial. In: The Lancet Oncology. Band 15, 2014, S. 966–977.
- EMA-Fachinformation
- SYLVANT™ (siltuximab) Receives FDA Approval to Treat Multicentric Castleman’s Disease (MCD)., PM von J&J, Janssen vom 23. April 2014, abgerufen am 29. April 2014.
- Mark Bower: How I treat HIV-associated multicentric Castleman disease. In: Blood. Band 116, Nr. 22, 25. November 2010, ISSN 0006-4971, S. 4415–4421, doi:10.1182/blood-2010-07-290213.
- E. Oksenhendler u. a.: Multicentric Castleman's disease in HIV infection: a clinical and pathological study of 20 patients. In: AIDS. 10/1/1996, S. 61–67. PMID 8924253
Weblinks
- M. Horger, R. Bares, J. Wiefels, W. Schöber, K. Müssig: Radiologische Befunde bei Morbus Castleman. In: RöFo – Fortschritte auf dem Gebiet der Röntgenstrahlen und der bildgebenden Verfahren. 179, 2007, S. 991, doi:10.1055/s-2007-990981.
- Detaillierte Informationen zum Thema Morbus Castlemann bei HIV Patienten