Hartnup-Krankheit
Die Hartnup-Krankheit (Hartnup-Syndrom) ist eine angeborene Störung des Transportes der meisten Aminosäuren durch die Körperzellen. Im Organismus ist dabei der Mangel der für den Menschen essentiellen Aminosäure Tryptophan am stärksten ausgeprägt.
| Klassifikation nach ICD-10 | |
|---|---|
| E72.0 | Störungen des Aminosäuretransportes |
| ICD-10 online (WHO-Version 2019) | |
| Klassifikation Hartnup-Krankheit | |
|---|---|
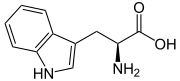 | |
| Tryptophan |
Epidemiologie
Die Krankheit wird autosomal-rezessiv vererbt. Mehrere Autoren, z. B. Tahmoush, Kimmig, berichten aber auch über leichtere Erkrankungen wie Lichtdermatosen und Aminosäureurie bei nahen Verwandten mit wahrscheinlich nur einem defekten Gen. Die Erstmanifestation ist im Kindesalter zwischen drei und neun Jahren. Sie kann auch früher[1] oder erst im Erwachsenen auftreten.[2][3]
Die Hartnup-Krankheit ist vergleichsweise selten und tritt mit einer Prävalenz von 1:24.000 auf. Bei weitem nicht alle Träger des entsprechenden Gendefekts entwickeln diese Krankheit. Auffallend ist der extreme Unterschied im Verlauf bei Personen, die durch Urin-Reihentests, und jenen, die erst durch einen akuten Ausbruch der Erkrankung gefunden wurden. Bei Ersteren wurden nur leichtere Erkrankungen beobachtet, viele erkrankten gar nicht. Bei Letzteren ist die Sterblichkeitsrate hoch.[1][2][4][5][6][7][8][9][10]
Bisher wurden 21 Mutationen von SLC6A19 in Chromosom 5 Genlocus p15.33 identifiziert, die den Aminosäuretransport unterschiedlich beeinträchtigen. Die meisten Betroffenen haben zwei unterschiedliche Mutationen. Das Allel D173N kommt bei 42 % der durch Urin-Screening bei Neugeborenen in Amerika, Australien und Kanada gefundenen Genträger vor.[11] Gemeinsamkeiten der D173N-Träger legen einen gemeinsamen Vorfahren vor mehr als tausend Jahren in Westeuropa nahe, etwa im heutigen Frankreich oder auf den Britischen Inseln. Fraglich bleibt, wie das Allel eine so hohe Heterozygotenfrequenz von circa einem von 122 Individuen erreichen konnte, da klare Hinweise auf einen Selektionsvorteil gesunder Anlageträger fehlen. Die Autoren postulieren, dass der lange Fortbestand dieses Allels auf seiner Unfähigkeit beruht, den Transport vollständig zu deaktivieren.[11] Bei manchen Mutationen wie den Allelen D173N und P265L ist die Anzahl der Transportkanäle abhängig von ACE2 im Darm bzw. Collectrin (Tmem27) in den Nieren.[12]
Die große Variabilität der Hartnup-Krankheit kann durch die Beteiligung von bis zu sechs genetischen Varianten erklärt werden: Je 1–2 Allele von SLC6A19 und je 2–4 Allele für die komplementäre Expression durch ACE2 und Collectrin. Dadurch wird alles von einer alleinigen und totalen Sperrung des Transports in den Nieren und vielen Zwischenstufen bis zu einer alleinigen und totalen Sperrung des Transports im Darm möglich. Auch das als harmlos geltende Allel D173N hat ohne die komplementäre Expression durch ACE2 und Collectrin ein letales Potential.[13]
Naturgemäß ist es bei der ersten Gruppe einfacher, Daten über eine relativ große Anzahl von Betroffenen zu erhalten, als Daten über die „wilden“ Fälle zu sammeln. Diese Daten beziehen sich jedoch auf Menschen, bei denen lebenslang ein erhöhter Bedarf an Vitaminen und Proteinen berücksichtigt wurde, was neben dem D173N-Allel eine mögliche Erklärung für die milden Verläufe bietet. Dies führte auch weltweit in Fachbüchern und Veröffentlichungen zu einer Darstellung der Hartnup-Krankheit als „benign“ (gutartig).
Pathogenese
Es handelt sich um eine Störung des Transportes von Aminosäuren durch die Membranen der Körperzellen. Dafür verantwortlich ist der Mangel eines bestimmten kanalartigen Membranproteins (neutraler Aminosäuretransporter), welches für die Durchschleusung von aromatischen und neutralen Aminosäuren durch die Zellen zuständig ist. Der Defekt ist allgegenwärtig, tritt aber in Geweben, die verstärkt für die Resorption (Aufnahme) von Aminosäuren konzipiert sind, deutlicher in Erscheinung. Die Störung betrifft hauptsächlich den oberen Dünndarmabschnitt (Jejunum) und die proximalen Tubuluszellen der Nieren, die den proximalen Tubulus bilden.
Durch das Unvermögen der Niere, bestimmte Aminosäuren im Blut zu halten (verminderte Rückresorption), sammeln sich diese im Harn und gehen dem Körper folglich verloren. Im Urin finden sich Alanin, Serin, Threonin, Valin, Leucin, Isoleucin, Phenylalanin, Tyrosin, Tryptophan, Glutaminsäure, Asparagin und Histidin. Vorausgesetzt, die Proteinaufnahme ist befriedigend, ist die im Urin ausgeschiedene Menge ernährungsphysiologisch unbedeutend. Wäre die Anomalie auf die Nieren beschränkt, würde sie keine Symptome verursachen und wäre kaum erkannt worden.[14]
Eine Resorptionsstörung dieser Aminosäuren im Dünndarm hat schwerwiegende Folgen. Ein Mangel dieser Substanzen im Körper ist eine Konsequenz dieser Resorptionsstörung. Der größte Teil der für den Lebenserhalt notwendigen essentiellen Aminosäuren wird durch Aminosäure-Recycling im Darm gewonnen.
Fortlaufend wird im Körper eine große Anzahl von Zellen abgebaut. Diese Zellen werden zerlegt und die Bestandteile – unter anderem Aminosäuren – in den Darm ausgeschieden. Bei der Hartnup-Krankheit gehen dabei die für den Körper wertvollen Aminosäuren durch die verminderte Rückresorption weitgehend verloren. Dadurch kann der Bedarf einer äußeren Zufuhr der von dem Transportdefekt betroffenen Aminosäuren stark ansteigen und ein Ausmaß erreichen, das durch bloße Nahrungsaufnahme nicht befriedigt werden kann. Auch normalerweise für Menschen nicht essentielle Aminosäuren können dadurch essentiell werden. Der menschliche Stoffwechsel kann fehlende Substanzen oftmals selbst herstellen und damit das Defizit auch weitgehend kompensieren. Problematisch erweist sich jedoch der Mangel an Tryptophan, einer essentiellen Aminosäure. Für dieses Molekül gibt es im menschlichen Stoffwechsel keine Möglichkeit zur Synthese. Ein Mangel an Tryptophan kann begrenzt durch eine Substitution mit dessen Folgeprodukten Niacin und Nicotinamid beseitigt werden. Individuell unterschiedlich werden 1/80 bis 1/40 des Tryptophans in Nicotinamid umgewandelt. Durch eine erhöhte Zufuhr von Nicotinamid muss der Körper weniger Tryptophan umwandeln, wodurch mehr für andere Zwecke zur Verfügung steht. Nicht resorbiertes Tryptophan wird im Darm durch Bakterien zu diversen Indolverbindungen metabolisiert. Diese werden im Gegensatz zum Tryptophan sehr wohl vom Darm resorbiert. Außerdem können sie die Blut-Hirn-Schranke überwinden. Viele zentralnervöse Symptome werden darauf zurückgeführt.
Symptomatik
Das Krankheitsbild gleicht im Wesentlichen dem der Pellagra (Niacinmangel). Bei Mangel an essentiellen Aminosäuren können auch die Symptome von Kwashiorkor auftreten. Auslösende Faktoren für einen Krankheitsschub sind Fieber oder einige fiebersenkende Medikamente, starke Sonnenexposition, Stress und bestimmte Medikamente (Sulfonamide), Inhibitoren des Vitamin-B-Komplexes, insbesondere von (Niacin), (Riboflavin) und (Pyridoxin).[15] Pellagra kann auch zu Störungen im Porphyrinstoffwechsel führen.[16] Porphyrieauslösende Medikamente können diese Störungen verstärken und schließlich einen Hartnup-Schub auslösen. Einige Mutationen können auch durch auf den Blutdruck wirkende Medikamente aktiviert werden (ACE2).
- Haut: Kennzeichnend für dieses Syndrom sind Hautläsionen in Form von erythematösen Ekzemen.
- Darm: Es kommt zu wiederkehrenden Durchfällen (Diarrhö).
- ZNS: Intermittierend finden sich neurologische oder psychiatrische Symptome. Es kann zu Störung der Bewegungskoordination (Ataxie), (Parkinsonoid), Lähmungen (Paresen) oder Stimmungsschwankungen kommen. Selten werden auch demenzähnliche Zustände, Schielen (Strabismus) und Krampfanfälle (Konvulsionen) beschrieben.
- Immunsystem: Mangelernährung schädigt das Immunsystem, Häufigkeit und Schwere von Infektionen nehmen zu.[17] Eine besondere Bedeutung für das Immunsystem haben L-Cystin und L-Cystein, welche deshalb Angriffsziele von HIV sind.[18] L-Cystin kann bei Hartnup essentiell werden.
- Leber, Nebennieren und Bauchspeicheldrüse: Pellagrakranke zeigen mit bemerkenswerter Regelmäßigkeit Überempfindlichkeit gegen Insulin. Zwei Organe, die Nebennieren und die Leber, beteiligt (mit anderen) in der Regulation des Kohlenhydrat-Stoffwechsels, zeigen schwere anatomische Schäden in den meisten Pellagrakranken.[19] Auch bei Hartnup wurden Schäden an diesen Organen berichtet.[10][20]
Diagnose
Jeder Patient, der an pellagraartigen Symptomen erkrankt und bei dem der für diese Krankheit typische ernährungsbedingte Niacinmangel nicht nachvollziehbar ist, sollte auf das Vorhandensein des Gendefekts für die Hartnup-Krankheit untersucht werden. Bei der relativ hohen Zahl an gefundenen Mutationen ist das aber sehr schwierig. In einer Studie[21] wurden nicht alle ursächlichen Allele in allen betroffenen Personen identifiziert, was die Möglichkeit erhöht, dass neben SLC6A19 andere Gene zur Hartnup-Krankheit beitragen könnten.
Die Diagnose der Hartnup-Krankheit kann meist mittels Urinanalyse durchgeführt werden. Man findet häufig eine neutrale Aminoazidurie (Vorhandensein von Aminosäuren im Harn). Durch eine nicht vorhandene Aminoazidurie kann die Hartnup-Krankheit jedoch nicht ausgeschlossen werden, wenn von einer ausreichenden Ernährung ausgegangen werden kann.[5][22][23][24][25] Es wurden auch zwei Fälle von Hypoaminoazidurie berichtet.[26]
Durch den Transportdefekt kann auch bei normalen Blutwerten ein Mangel in den Zellen bestehen. Paradoxerweise können durch die eingeschränkte Aufnahmefähigkeit der Zellen die Blutwerte der betroffenen Aminosäuren sogar erhöht sein, wenn auch bei weitem nicht so stark wie z. B. bei Hypertryptophanämie.
Bei fehlender Aminoazidurie und scheinbar normalen Blutwerten kann ein einfacher Test auf Porphyrogene (Indolylacrylsäure und nahe Verwandte, Absorptionsmaximum von 509 µm) einen weiteren Hinweis geben.[27] Dabei wird in den Morgenurin etwas Salzsäure geben (pH = 2 bis 2,5) und die Probe erwärmt. Vorhandene Porphyrogene bilden dann einen roten Farbstoff, durch den sich der Urin nahezu schwarz verfärben kann. Dieser Test kann auch bei Porphyrie und einigen anderen Erkrankungen sowie nach Einnahme von Phenothiazinderivaten positiv sein.
Referenzbereich Tryptophan im Plasma oder Serum:[28]
- Säuglinge: 25 µmol/l bis 69 µmol/l (0,5 mg/dl bis 1,3 mg/dl)
- Kinder: 32 µmol/l bis 79 µmol/l (0,6 mg/dl bis 1,6 mg/dl)
- Erwachsene: 34 µmol/l bis 90 µmol/l (0,7 mg/dl bis 1,7 mg/dl)
Von einigen Laboratorien werden irrtümlich extrem niedrige untere Grenzwerte, bis hinunter zu 10 µmol/l, angegeben, die unter Umständen schon letal sein können.[29]
Therapie
Die Behandlung besteht in der Verabreichung der fehlenden Substanzen (Substitutionstherapie). Eine tägliche Nahrungsergänzung von 50 bis 250 mg Nicotinamid verbessert die Situation meist bedeutend, manchmal bewirkt es nur wenig[29] oder bleibt wirkungslos.[24] Aufgrund der Lebertoxizität von Nicotinamid wird jedoch besser Niacin eingenommen. Tägliche Dosen bis 3 Gramm Niacin in einer Erstphase und danach 500 mg pro Tag in der Erhaltungsphase sind ideal. Das bei Niacin entstehende Flushing wird zwar oft als unangenehm empfunden, geht jedoch nach ein paar Wochen wieder weg.
Üblicherweise wird eine proteinreiche Diät mit hohem Anteil an Tryptophan empfohlen. Tryptophan findet sich hauptsächlich in Milch und Milchprodukten sowie in Geflügel, Rindfleisch, Nüssen und Kartoffeln.
In schweren Fällen ist es nicht möglich, den Blutspiegel der von dem Transportdefekt betroffenen Aminosäuren durch eine mit Proteinen angereicherte Diät signifikant zu erhöhen. Der Mangel muss dann durch Umgehung des Transportdefekts mittels intravenöser Substitution oder oraler Substitution mit chemisch abgewandelten Aminosäuren beseitigt werden.[29] Geschieht dies nicht, so führt der Mangel mittelfristig zu schweren Schäden und langfristig zum Tod.
Die Photodermatosen sind mit einer harnstoffhaltigen (10 bis 15 %) Salbe gut behandelbar, auch bei Blasenbildung.
Historisches
Im Jahr 1955 wurde von Hersov der erste bekannte Fall wissenschaftlich beschrieben: ein 10-jähriger Junge mit psychotischer Neigung und Dermatose, jedoch ohne eruierbare Pellagra, sprach gut auf die Gabe von Nikotinamid an. Ausführlich beschrieben wurde die Krankheit schließlich 1956 in London bei einer konsanguinen Familie namens Hartnup, bei welcher vier von acht Kindern Symptome der Krankheit zeigten und nach welcher die Krankheit schließlich benannt wurde.[30][31] Später konnte bei dieser Familie die Splice-Variante IVS8+2G festgestellt werden, die mit einem völligen Funktionsverlust des Proteins einhergeht und später auch in compound-heterozygoten Familien in Australien bestätigt werden konnte.[11] Möglicherweise litt auch die julisch-claudische Imperatorenfamilie von Julius Caesar bis hin zu Kaiser Nero an dieser Krankheit.[32]
Literatur
- Dennis L. Kasper (Hrsg.), Eugene Braunwald (Hrsg.), Anthony Fauci: Harrison’s Principles of Internal Medicine. 16th Edition Volume II ISBN 0-07-139142-8
Einzelnachweise
- K. Schmidtke, W. Endres u. a.: Hartnup syndrome, progressive encephalopathy and allo-albuminaemia. A clinico-pathological case study. In: European Journal of Pediatrics. Band 151, Nummer 12, Dezember 1992, S. 899–903, ISSN 0340-6199. PMID 1473543.
- E. Mori, A. Yamadori u. a.: [Adult-onset Hartnup disease presenting with neuropsychiatric symptoms but without skin lesions]. In: Rinsho shinkeigaku = Clinical neurology. Band 29, Nummer 6, Juni 1989, S. 687–692, ISSN 0009-918X. PMID 2582682. (Artikel in Japanisch)
- A. Oakley, J. Wallace: Hartnup disease presenting in an adult. In: Clinical and Experimental Dermatology. Band 19, Nummer 5, September 1994, S. 407–408, ISSN 0307-6938. PMID 7955499.
- K. H. Daute, K. Dietel, W. Ebert: Das Hartnupsyndrom – Bericht über einen tödlichen Krankheitsverlauf. In: Zeitschrift für Kinderheilkunde. Band 95, Nummer 2, Januar 1966, S. 103–113, ISSN 0044-2917. PMID 5983075.
- A. J. Tahmoush, D. H. Alpers u. a.: Hartnup disease. Clinical, pathological, and biochemical observations. In: Archives of Neurology. Band 33, Nummer 12, Dezember 1976, S. 797–807, ISSN 0003-9942. PMID 999542.
- D. Milovanović, A. Djukić u. a.: [Hartnup disease (report of 2 cases in one family)]. In: Srpski arhiv za celokupno lekarstvo. Band 128, Nummer 3–4, 2000 Mar-Apr, S. 97–103, ISSN 0370-8179. PMID 10932618. (Artikel in Serbisch)
- OMIM, Hartnup Disorder
- Kazuhiko Oyanagi, Masayo Takagi, Michiyuki Kitabatake and Tooru Nakao. Hartnup Disease, Tohoku J. exp. Mod.,1967, 91, 383-395
- Examples of autopsy reports – Non coroner, Michael Fishbein, MD (March 08)
- Hartnup disease, By M. D. MILNE. Medical Unit, Westminster, Medical School, London, S.W. 1
- D. N. Azmanov, H. Rodgers u. a.: Persistence of the common Hartnup disease D173N allele in populations of European origin. In: Annals of Human Genetics. Band 71, Pt 6 November 2007, S. 755–761, ISSN 0003-4800. doi:10.1111/j.1469-1809.2007.00375.x. PMID 17555458.
- S. M. Camargo, D. Singer u. a.: Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. In: Gastroenterology. Band 136, Nummer 3, März 2009, S. 872–882, ISSN 1528-0012. doi:10.1053/j.gastro.2008.10.055. PMID 19185582.
- Critical Review: The role of the neutral amino acid transporter B0AT1 (SLC6A19) in Hartnup disorder and protein nutrition. https://iubmb.onlinelibrary.wiley.com/doi/full/10.1002/iub.210#sec1-6
- Br Med J. 1964 February 8; 1(5379): 327–336. Disorders of Amino-Acid Transport* M. D. Milne
- Vitamin-Lexikon: Bässler, Grühn, Loew, Pietrzik
- Pschyrembel® Klinisches Wörterbuch Ausgabe 253
- Z. Orbak, V. Ertekin u. a.: Hartnup disease masked by kwashiorkor. In: Journal of health, population, and nutrition. Band 28, Nummer 4, August 2010, S. 413–415, ISSN 1606-0997. PMID 20824986. PMC 2965334 (freier Volltext).
- HIV-induced cysteine deficiency and T-cell dysfunction — a rationale for treatment with N-acetylcysteine. W. Dröge, H.-P. Eck, S. Mihm
- Insulin-hypersensitivity in Pellagrins and its significance, Fr. Mainzer, Anglo-Swiss Hospital, Alexandria, Egypt.
- omim.org entry 234500
- H. F. Seow, S. Bröer u. a.: Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. In: Nature genetics. Band 36, Nummer 9, September 2004, S. 1003–1007, ISSN 1061-4036. doi:10.1038/ng1406. PMID 15286788.
- E. Freundlich, M. Statter, S. Yatziv: Familial pellagra-like skin rash with neurological manifestations. In: Archives of Disease in Childhood. Band 56, Nummer 2, Februar 1981, S. 146–148, ISSN 1468-2044. PMID 6451201. PMC 1627116 (freier Volltext).
- E. R. Da Gloria, J. G. Assunção, M. A. Costa: [Clinical picture of Hartnup disease. Without urine amino acids or any other identified metabolic disorder (a new entity)]. In: Medicina cutánea ibero-latino-americana. Band 18, Nummer 4, 1990, S. 227–231, ISSN 0210-5187. PMID 2077308. (Artikel auf Portugiesisch)
- P. F. Borrie, C. A. Lewis: Hartnup disease. In: Proceedings of the Royal Society of Medicine. Band 55, März 1962, S. 231–232, ISSN 0035-9157. PMID 13871450. PMC 1896568 (freier Volltext).
- M. Goulon, R. Escourolle u. a.: Pellagre endogène, sans hyperaminoacidurie. In: Revue neurologique. Band 120, Nummer 3, März 1969, S. 149–158, ISSN 0035-3787. PMID 5343986. (Artikel in Französisch)
- Voiculescu V. · Luca N. · Hategan D. Biochemical Research in Two Cases of Endogenous Pellagra. Eur Neurol 1973;10:205–214 (doi:10.1159/000114277)
- J. Kimmig-Hamburg: Stoffwechselstörungen bei polymorphen Lichtdermatosen und neuere Untersuchungsergebnisse im Zusammenhang mit dem Hartnup-Syndrom. In: Archiv für Klinische und Experimentelle Dermatologie. Band 219, Nummer 1, 1964, S. 753–762. ISSN 1432-069X doi:10.1007/BF00522954
- Lehrbuch der Klinischen Chemie und Pathobiochemie, Schattauer 3. Auflage
- A. J. Jonas, I. J. Butler: Circumvention of defective neutral amino acid transport in Hartnup disease using tryptophan ethyl ester. In: The Journal of clinical investigation. Band 84, Nummer 1, Juli 1989, S. 200–204, ISSN 0021-9738. doi:10.1172/JCI114141. PMID 2472426. PMC 303970 (freier Volltext).
- D. N. Baron, C. E. Dent u. a.: Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria, and other bizarre biochemical features. In: Lancet. Band 271, Nummer 6940, September 1956, S. 421–428, ISSN 0140-6736. PMID 13358233.
- Hersov and Rodnight, 1960, J. Neurol. Neurosurg. Psychiat., 1960, 23, 40, Link: https://jnnp.bmj.com/content/jnnp/23/1/40.full.pdf
- J. H. Dirckx: Julius Caesar and the Julian emperors. A family cluster with Hartnup disease? In: The American Journal of Dermatopathology. Band 8, Nummer 4, August 1986, S. 351–357, ISSN 0193-1091. PMID 3532855.
Weblinks
- Hartnup-Krankheit. In: Online Mendelian Inheritance in Man. (englisch)
- Hartnup-Krankheit. In: Orphanet (Datenbank für seltene Krankheiten).