Hämoglobin-Elektrophorese
Die Hämoglobin-Elektrophorese (Hb-Elektrophorese) ist eine der wichtigsten Untersuchungsmethoden zur Abklärung von Hämoglobinopathien.[1] Unter diesem Begriff wird die Auftrennung der Hämoglobine mit unterschiedlichen elektrophoretischen Methoden wie z. B. der Gelelektrophorese, der Kapillarelektrophorese (CE) oder der isoelektrischen Fokussierung zusammengefasst. Auch die HPLC wird häufig der Hämoglobin-Elektrophorese zugerechnet, obwohl die Auftrennung hierbei chromatographisch erfolgt. Untersucht wird ein Hämolysat, in dem die Hämoglobine frei in Lösung vorliegen und nicht mehr in den Erythrozyten eingeschlossen sind.
Strukturvarianten
Insbesondere zur Abklärung von Hämoglobinstrukturvarianten ist die Hämoglobin-Elektrophorese gut geeignet. Bei diesen Strukturanomalien liegen auf Grundlage einer veränderten Aminosäuresequenz Hämoglobine mit neuen Eigenschaften vor. Bei vielen dieser Anomalien ändert sich durch die Sequenzvariante die Säurekonstante des Proteins, sodass sich bei gegebenem pH-Wert die Gesamtladung des Moleküls verändert, was die Wanderung im elektrischen Feld bzw. die Interaktion mit den Phasen der HPLC beeinflusst. Beispiele mit deutlicher Veränderung der Säure-Baseeigenschaften sind Hämoglobin C (HbC) und E (HbE), bei denen die saure Glutaminsäure durch das basische Lysin ersetzt wird.[2]
Dennoch lassen sich je nach Methode nicht alle Varianten trennscharf voneinander abgrenzen, da manche Hämoglobinvarianten bei einem bestimmten pH-Wert sehr ähnliche Wanderungseigenschaften aufweisen. Standardverfahren zu initialen Untersuchung ist eine Hb-Elektrophorese in alkalischem Milieu. Ergibt sich in dieser Untersuchung ein Hinweis auf das Vorliegen einer Strukturvariante, die nicht eindeutig spezifiziert werden kann, ist anschließend eine Untersuchung in saurem Milieu möglich. Strukturanomalien, die in der alkalischen Elektrophorese ein identisches Migrationsmuster aufweisen, können bei hoher Protonenkonzentration andere Ladungseigenschaften aufweisen und daher in der sauren Elektrophorese voneinander abgegrenzt werden. So komigrieren bspw. HbC und HbE im alkalischen Milieu und können bei saurem Milieu deutlicher voneinander unterscheiden werden. Bei verschiedenen anomalen Hämoglobinen ist eine Trennung mittels elektrophoretischer Methoden nicht möglich.[3]
Wichtig ist eine zuverlässige Quantifizierung der einzelnen vorliegenden Hämoglobin-Entitäten, die üblicherweise mittels CE oder HPLC erfolgt. Der Anteil einer gefundenen Strukturvariante und das Verhältnis der einzelnen Fraktionen zueinander kann einen Hinweis auf eine (möglicherweise zusätzlich vorliegende) Hämoglobinopathie aus dem Thalassämiespektrum geben.
Thalassämien
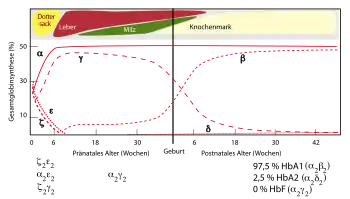
Zur Diagnostik einer β-Thalassämie ist eine valide Quantifizierung der Hämoglobinfraktionen unerlässlich: Bei den Thalassämien entstehen keine Hämoglobine mit veränderten Eigenschaften, sondern die Produktion der physiologischen Hämoglobine ist bisweilen erheblich beeinträchtigt. Das Hämoglobin des Erwachsenen besteht zum überwiegenden Teil aus dem Hämoglobin A (HbA, ~95%), dem Hämoglobin A2 (HbA2, <3,5 %) sowie in Spuren aus dem fetalen Hämoglobin F (HbF, <0,5 %). HbA besteht aus einem Tetramer aus zwei α- und zwei β-Globinketten (α2β2), während HbA2 aus zwei α- und zwei δ-Globinketten (α2δ2) gebildet wird. HbF entsteht aus zwei α- und zwei γ-Ketten (α2γ2). Bei den β-Thalassämien ist die Produktion der β-Globine vermindert.[4] Dies führt zu einer relativen Vermehrung des HbA2 und bisweilen des HbF, die quantifiziert werden kann. Bei der β-Thalassämia minor liegt der HbA2-Anteil üblicherweise bei >3,5 %. Bei der β-Thalassämia major findet sich überwiegend HbF.[5]
Die Diagnose der α-Thalassämien hingegen ist mit der Hb-Elektrophorese nur eingeschränkt möglich. Da die α-Globine an allen fetalen und adulten Hämoglobinen beteiligt sind, kommt es nicht zu einer relativen Zunahme einer Fraktion, die diagnostisch verwertbar wäre. Bei den α-Thalassämien entsteht je nach Krankheitsbild durch den Überschuss der β-Ketten ein sehr instabiles β-Globintetramer, das HbH (β4). U. a. aufgrund seiner Instabilität entgeht es häufig einem laborchemischen Nachweis in der Routinediagnostik. Bei der HbH-Krankheit finden sich jedoch bisweilen Mengen, die nachgewiesen werden können. α-Thalassämien werden daher üblicherweise mittels Hämoglobin-Genotypisierung des HBA-Lokus untersucht. Eine Hb-Genotypisierung bietet sich auch zur molekulargenetischen Sicherung von Hämoglobinstrukturvarianten, zur Abklärung unklarer Fälle oder der weiteren Aufarbeitungen von komplexen (z. B. compound heterozygoten) Hämoglobinopathie-Kombinationsformen an.[6]
Befundung
Bei der Bewertung der Befunde einer Hb-Elektrophorese ist die Berücksichtigung der übrigen Befunde sowie der Klinik und Anamnese des Patienten von großer Bedeutung. Eine Kenntnis der Parameter des roten Blutbildes ist hierbei unerlässlich. So muss der elektrophoretische Befund mit dem Blutbild gemeinsam befundet werden. Hämoglobinbefund und Erythrozytenparameter sollten zueinander passen. Hierbei auftretende Diskrepanzen sollten an weitere, zusätzlich vorliegende Pathologien denken lassen. Bei entsprechendem Verdacht kann dann eine weitergehende Diagnostik veranlasst werden. Häufigstes Beispiel für eine solche Konstellation sind die α-Thalassämien: Bei hinweisenden Blutbildparametern (z. B. Erythrozytose, Hypochromasie, Mikrozytose) findet sich eine unauffällige Hb-Elektrophorese. Das Blutbild sollte dann nach Ausschluss häufiger Ursachen für ähnliche Veränderungen (v. a. Fe-Mangel) und unter Berücksichtigung der Anamnese (z. B. ethnische Herkunft) Anlass für eine weitere Abklärung z. B. mittels Hb-Genotypisierung sein.[7]
Zu berücksichtigen ist bei der Auswertung auch das Alter des Patienten. Erst kurz vor der Geburt erfolgt eine Umstellung vom fetalen auf das adulte Hämoglobin. Daher lassen sich bei Säuglingen noch physiologischerweise große Mengen an HbF nachweisen. Aber auch sämtliche Pathologien, die die β-Globinkette betreffen, bilden sich durch die allmählich zunehmende β-Globinsynthese erst vollständig in den ersten Lebensmonaten aus.
Einzelnachweise
- Ronald J. A. Trent: Diagnosis of the haemoglobinopathies. In: The Clinical Biochemist. Reviews. Band 27, Nr. 1, Februar 2006, ISSN 0159-8090, S. 27–38, PMID 16886045, PMC 1390791 (freier Volltext).
- Enno Kleihauer unter Mitarbeit von Elisabeth Kohne und Andreas E. Kulozik: Anomale Hämoglobine und Thalassämiesyndrome : Grundlagen und Klinik. Ecomed, Landsberg 1996, ISBN 3-609-62760-3.
- Hämoglobindiagnostik | Universitätsklinikum Freiburg. Abgerufen am 10. April 2020.
- Beta Thalassämie. Abgerufen am 10. April 2020.
- G. M. Clarke, T. N. Higgins: Laboratory investigation of hemoglobinopathies and thalassemias: review and update. In: Clinical Chemistry. Band 46, 8 Pt 2, August 2000, ISSN 0009-9147, S. 1284–1290, PMID 10926923.
- S1-Leitlinie Thalassämie AWMF 025/017. (PDF) 30. Juni 2016, abgerufen am 10. April 2020.
- Stufendiagnostik bei Verdacht auf eine Hämoglobinopathie. (PDF) Labor 28 Berlin, Januar 2018, abgerufen am 10. April 2020.