Hämoglobin C
Hämoglobin C (Hb C oder HbC, kristallines Hb) ist eine Hämoglobin-Strukturvariante und gehört somit zu den erblich-bedingten Hämoglobinopathien. Verursacht wird sie durch eine Punktmutation im HBB-Gen, das für die β-Globin-Kette des Hämoglobins codiert. Durch die HBB-Mutation c.19G>A findet sich im β-Globin an der Aminosäureposition 6 anstelle der sauren Glutaminsäure ein basischer Lysinrest (E6K-Substitution).[1]
| Klassifikation nach ICD-10 | |
|---|---|
| D58.2 | Sonstige Hämoglobinopathien
HbC-Heterozygotie/-Homozygotie |
| D57.2 | Doppelt heterozygote Sichelzellenkrankheiten
HbSC-Krankheit |
| D56.8 | Sonstige Thalassämien
HbC-β-Thalassämie |
| ICD-10 online (WHO-Version 2019) | |
Klinische Bedeutung
Bei der klinischen Relevanz muss zwischen der HbC-Trägerschaft oder -Heterozygotie und der HbC-Homozygotie unterschieden werden. Bei letzterer weisen beide HBB-Gene die HbC-Mutation auf. Patienten mit einer HbC-Homozygotie haben jeweils von Mutter und Vater ein mutiertes Allel geerbt. Dadurch besteht bei diesen das Hämoglobin überwiegend (>95 %) aus Hämoglobin C. Durch die gegenüber dem physiologischen Hämoglobin A (HbA) reduzierte Löslichkeit des HbC kommt es zur Bildung von HbC-Kristallen in den Erythrozyten. Diese Zellen werden in der Milz sequestriert. HbC-Homozygote weisen durch den Abbau der Erythrozyten eine meist milde hämolytische Anämie auf. Im Blutausstrich imponieren neben Targetzellen Mikrosphärozyten und Kristallzellen.[2]
Bei der HbC-Trägerschaft weist ein HBB-Allel keine Mutation auf und die Träger bilden daher neben HbC auch das physiologische HbA. Diese Anlageträger sind klinisch stumm – zeigen also keinerlei Symptome. Lediglich leichte zelluläre Veränderung der Erythrozyten können z. B. in Form von Targetzellen beobachtet werden. HbC-Träger vererben jedoch das HbC-Allel an die Hälfte ihrer Nachkommen, sodass sich je nach möglicherweise beim Partner vorliegenden Mutationen klinisch relevante Kombinationsformen ergeben können. In Ländern mit einer hohen Allelfrequenz bestehen daher entsprechende Screening-Programme, um auch asymptomatische Träger z. B. zur Familienplanung zu erkennen.[3]
Ein erheblicher Krankheitswert ergibt sich jedoch aus Kombinationen mit anderen Hämoglobinopathien. Besondere Bedeutung haben hierbei compound heterozygote Konstellationen mit Strukturvarianten der β-Kette und β-Thalassämien.
Die häufigste und klinisch relevanteste ist die Kombination aus dem „Sichelzell-Allel“, auch HbS genannt, und HbC. Diese so genannte HbSC-Krankheit ist v. a. in Afrika häufig, da sich in Westafrika die Verbreitungsgebiete von HbS und HbC überschneiden. Das klinische Erscheinungsbild entspricht einer klinischen abgemilderten Sichelzellkrankheit, da HbC nicht so leicht wie HbS polymerisiert. Es gibt bei der HbSC-Krankheit daher z. B. weniger akute Gefäßverschlüsse. Andererseits weisen HbSC-Patienten häufiger signifikante Retinopathien, avaskuläre Nekrosen des Knochens oder einen Priapismus auf.[4][5] Morphologisch zeigen sich im Ausstrich Targetzellen. Die für die Sichelzellkrankheit charakteristischen Sichelzellen finden sich bei der HbSC-Krankheit eher selten. Dafür finden sich s.g. Kristallzellen, die bei der Sichelzellkrankheit nicht auftreten.
Bei der HbC-βo-Thalassämie hat der Patient neben dem HbC-Allel auch ein HBB-Allel geerbt, das kein β-Globin produziert (βo-Allel). Daher bilden diese Patienten wie bei der HbC-Homozygotie überwiegend Hämoglobin C. Im Gegensatz hierzu ist aber nur ein funktionelles HBB-Gen vorhanden, sodass sich zusätzlich zur leichten Hämolyse die klassischen Merkmale einer β-Thalassämia minor mit deutlicher Mikrozytose und Hypochromasie gesellen.[6]
Epidemiologie
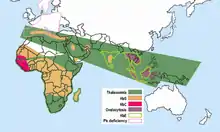
Die ursächliche Mutation entstand in Westafrika. Da Hämoglobin C einen relativen Schutz gegenüber der in dieser Region häufigen Malaria verleiht, bot die HbC-Trägerschaft einen Selektionsvorteil.[7] Dies führte dazu, dass in Westafrika bis zu 25 % der Bewohner HbC-Träger sind oder eine HbC-Homozygotie aufweisen.[8] Aus diesen Gebieten gelangte die Mutation durch den massenhaften Sklavenhandel v. a. nach Amerika und in die Karibik. Als Folge weisen 2–3 % der Afroamerikaner in den USA das HbC-Allel auf. Durch den Sklavenhandel ist das HbS-Allel in den USA ebenfalls relativ häufig bei den heutigen Nachfahren der ehemaligen Sklaven zu finden. Dadurch tritt in den USA die HbSC-Erkrankung signifikant häufiger als die HbC-Homozygotie auf.[9] HbC ist nach Hämoglobin E und HbS die weltweit dritthäufigste Hb-Strukturanomalie. In Deutschland kommt HbC hingegen relativ selten vor. Auch klinisch relevante Kombinationsformen sind daher entsprechend selten.[10]
Einzelnachweise
- HbVar ID 227. Abgerufen am 21. Juli 2020.
- Enno Kleihauer unter Mitarbeit von Elisabeth Kohne und Andreas E. Kulozik: Anomale Hämoglobine und Thalassämiesyndrome : Grundlagen und Klinik. Ecomed, Landsberg 1996, ISBN 3-609-62760-3.
- Sickle cell and thalassaemia (SCT) screening: programme overview. Abgerufen am 3. April 2020 (englisch).
- Lydia H. Pecker, Beverly A. Schaefer, Lori Luchtman-Jones: Knowledge insufficient: the management of haemoglobin SC disease. In: British Journal of Haematology. Band 176, Nr. 4, Februar 2017, S. 515–526, doi:10.1111/bjh.14444, PMID 27982424, PMC 5303157 (freier Volltext) – (wiley.com [abgerufen am 3. April 2020]).
- R. L. Nagel, M. E. Fabry, M. H. Steinberg: The paradox of hemoglobin SC disease. In: Blood Rev. Band 17, Nr. 3, September 2003, S. 167–178, doi:10.1016/S0268-960X(03)00003-1, PMID 12818227.
- Orphanet: Hemoglobin C beta thalassemia syndrome. Abgerufen am 3. April 2020.
- Steinberg, Martin H.: Disorders of hemoglobin : genetics, pathophysiology, and clinical management. 2nd ed Auflage. Cambridge University Press, New York 2009, ISBN 978-0-521-87519-6.
- Mark A. Travassos, Drissa Coulibaly, Matthew B. Laurens, Ahmadou Dembélé, Youssouf Tolo: Hemoglobin C Trait Provides Protection From Clinical Falciparum Malaria in Malian Children. In: Journal of Infectious Diseases. Band 212, Nr. 11, 1. Dezember 2015, ISSN 0022-1899, S. 1778–1786, doi:10.1093/infdis/jiv308, PMID 26019283, PMC 4633765 (freier Volltext).
- R. M. Fairhurst, H. Fujioka, K. Hayton, K. F. Collins, T. E. Wellems: Aberrant development of Plasmodium falciparum in hemoglobin CC red cells: implications for the malaria protective effect of the homozygous state. In: Blood. Band 101, Nr. 8, April 2003, S. 3309–3315, doi:10.1182/blood-2002-10-3105, PMID 12480691.
- Elisabeth Kohne, Enno Kleihauer: Hemoglobinopathies in Germany. In: Deutsches Aerzteblatt Online. 5. Februar 2010, ISSN 1866-0452, doi:10.3238/arztebl.2010.0065, PMID 20186311, PMC 2828242 (freier Volltext).