Pulvinon
Das Pulvinon ist eine organische chemische Verbindung, die zu den Lactonen, Estern, Enolen und zur Gruppe der Pulvinsäure-Farbstoffe zählt. Obwohl das Pulvinon kein Naturstoff ist, sind verschiedene natürlich vorkommende hydroxylierte Derivate bekannt, die von Pilzen, wie etwa dem Goldröhrling (Boletus elegans, auch als Suillus grevillei bezeichnet), oder von Schimmelpilzen wie Aspergillus Terreus produziert werden. Im Vergleich zur Vulpinsäure und zur Pulvinsäure besitzt das Pulvinon eine geringe Toxizität.
| Strukturformel | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||
| (E)-Form (links) und (Z)-Form (rechts) | |||||||||||||
| Allgemeines | |||||||||||||
| Name | Pulvinon | ||||||||||||
| Andere Namen |
5-Phenylmethylen-4-hydroxy-3-phenylfuran-2(5H)-on (IUPAC) | ||||||||||||
| Summenformel | C17H12O3 | ||||||||||||
| Externe Identifikatoren/Datenbanken | |||||||||||||
| |||||||||||||
| Eigenschaften | |||||||||||||
| Molare Masse | 322,33 g·mol−1 | ||||||||||||
| Aggregatzustand |
fest | ||||||||||||
| Schmelzpunkt | |||||||||||||
| Sicherheitshinweise | |||||||||||||
| |||||||||||||
| Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. | |||||||||||||
Geschichte und Vorkommen
In Pilzen wie den Dickröhrlingsverwandten und Flechten kommen sehr viele verschiedene Farbstoffe vor, die entweder Derivate der Pulvinsäure (etwa die Gomphidsäure) oder aus mehreren Pulvinsäure-Einheiten zusammengesetzt sind (Beispiel für zwei Einheiten: Badione). 1831 wurde der Pulvinsäure-Methylester Vulpinsäure bei der Untersuchung von Flechten (Cetraria Vulpina) durch den französischen Apotheker und Chemiker Antoine Bebert entdeckt, aber erst 1860 von Franz Möller und Adolph Strecker genauer untersucht und beschrieben.[3] Bei dem Versuch, die Struktur der Vulpinsäure zu klären, fand Adolf Spiegel[4][5][6][7] im Jahr 1880, dass die Vulpinsäure zu einer Disäure verseift werden konnte. Die erhaltene Disäure nannte er Pulvinsäure. 1894 wurde die Konstitution der Pulvinsäure von Jacob Volhard[8] geklärt, indem er Pulvinsäure durch basische Hydrolyse eines entsprechenden Dinitrils synthetisch herstellen konnte. Dabei erhielt er auch geringe Mengen eines Nebenprodukts, welches ein Jahr später von Ludwig Claisen und Th. Ewan[9] separat dargestellt und als 5-Benzyliden-4-hydroxy-3-phenylfuran-2(5H)-on charakterisiert wurde.
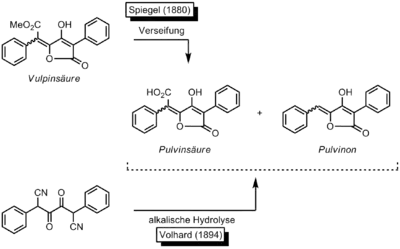
Claisen und Ewan beschrieben es als das der Pulvinsäure zu Grunde liegende Lacton: So entstand kurz danach der Name Pulvinon.
Biologische Bedeutung
Interessanterweise wurde die Bezeichnung Pulvinon erst ein Jahrhundert nach der Entdeckung des Urpulvinons zu einem Sammelbegriff, nämlich als 1973 von Edwards und Gill das erste natürliche hydroxylierte Derivat isoliert wurde.[10] Dabei handelte es sich um ein trihydroxyliertes Pulvinon, das als gelbes Pigment im europäischen Pilz Goldröhrling (Boletus elegans, auch als Suillus grevillei bezeichnet) gefunden wurde. Im selben Jahr 1973 wurden auch von Seto und seinen Mitarbeitern in Kulturen des Schimmelpilzes Aspergillus terreus Pulvinone gefunden.[11][12][13] Um diese von dem aus Suillus grevillei isolierten Naturstoff zu unterscheiden, wurden diese Verbindungen Aspulvinone genannt[14] und nach der chromatographischen Elutionsreihenfolge alphabetisch benannt.[15][16][17] (Das unpolarste Aspulvinon wurde somit zum Aspulvinon A, das am nächsten eluierende zum Aspulvinon B etc.).
Wie viele andere gelbe Farbstoffe in Pilzen und Flechten leitet sich das Pulvinon von der Pulvinsäure ab. Damit ist das Pulvinon-Strukturmotiv in vielen Naturstoffen vertreten. Alle Derivate des Pulvinsäuremonomers wie die Pulvinsäure selbst und etwa die Gomphidsäure, die Vulpinsäure, die Aspulvinone, und Derivate des Pulvinsäuredimers wie z. B. die Badione,[18] Norbadione[19][20] oder des Pulvinondimers wie das Aurantricholon[21] beinhalten dieses Motiv. In der Regel sind alle natürlich vorkommenden Pulvinonderivate (Z)-konfiguriert.

Biosynthese
In Pilzen verläuft die Biosynthese über aromatische Aminosäuren wie Phenylalanin und Tyrosin; daraus entsteht nach Oxidesaminierung zur Arylbrenztraubensäure, Dimerisierung, Ringspaltung und Decarboxylierung schließlich das Pulvinongerüst.[22]
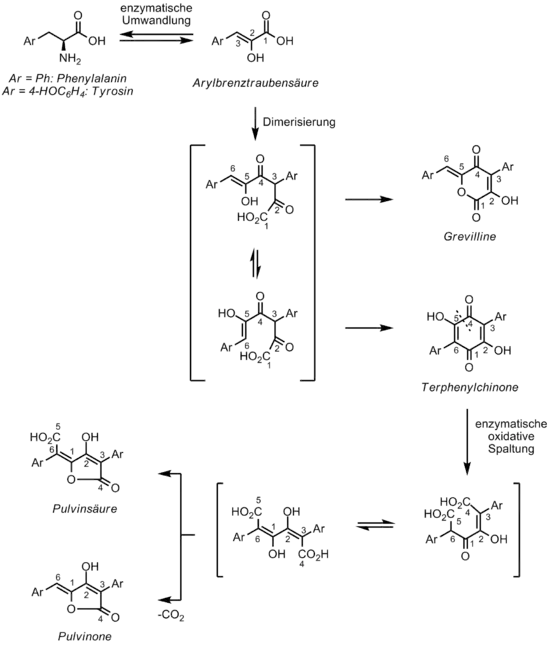
Chemische Eigenschaften
Das Pulvinon ist ein Lacton, also ein intramolekularer Ester der trans-1,4-Diphenyl-2,3-dihydroxy-1,3-butadien-1-carbonsäure und entsteht aus dieser durch Abspaltung von Wasser:

Pharmakologische Aktivität
- Rehse et al[23][24][25][26] demonstrierten, dass Vertreter der Familie der Pulvinone eine gerinnungshemmende Aktivität bei Ratten zeigen.
- Anfang der 80er Jahre wurden von den Firmen ICI und Smith Kline & French viele Derivate der Vulpinsäure wegen entzündungshemmender, antipyretischer und schmerzstillender Eigenschaften patentiert. Allerdings ist Vulpinsäure, genauso wie viele seine Derivate, ein Zellgift. Da Pulvinone eine geringere Zytotoxizität im Vergleich zu den entsprechenden Vulpinsäurederivaten zeigten, wurde das entzündungshemmende Potenzial von mehr als 100 Pulvinonen von der Firma Organon untersucht.[27] Bis heute ist nur wenig über die Ergebnisse dieser Untersuchung bekannt.
- Die Firma Wyeth patentierte 2005 biphenylsubstituierte Pulvinone[28][29] wegen ihrer sehr guten Aktivität gegen Gram-positive Bakterien. Allerdings wurden auf dem Pulvinongerüst basierende Antibiotika bis jetzt nur bei Tieren eingesetzt.
Darstellung
Jacob Volhard synthetisierte als erster die Vulpinsäure, die Pulvinsäure und das Pulvinon.[8] Bis heute wurden zehn Totalsynthesen von Pulvinonen veröffentlicht:
- 1895 von Claisen und Ewan,[9]
- 1975 und 1979 von Knight und Pattenden,[30][31]
- 1979 von Jerris, Wovkulich und Amos B. Smith III,[32]
- 1984 von Ramage et al.,[1]
- 1985 von Campbell et al.,[27]
- 1990 von Gill et al.,[33]
- 2005 von Caufield et al.,[28]
- 2006 von Antane et al.,[29]
- 2007 von Kaczybura und Brückner,[34]
- 2007 von Bernier, Moser und Brückner.[35][36]
Einzelnachweise
- R. Ramage, G. J. Griffiths, F. E. Shutt, J. N. A. Sweeney, J. Chem. Soc., Perkin Trans. 1, 1984, 1539–1545. doi:10.1039/P19840001539.
- Dieser Stoff wurde in Bezug auf seine Gefährlichkeit entweder noch nicht eingestuft oder eine verlässliche und zitierfähige Quelle hierzu wurde noch nicht gefunden.
- Canstatt’s Jahresbericht über die Fortschritte in der Pharmacie und verwandte Wissenschaften in allen Ländern, Harvard-Universität, Jahrgang 10 (1861).
- A. Spiegel, Ber. Dtsch. Chem. Ges., 1880, 13, 2, 1629–1635. doi:10.1002/cber.18800130293.
- A. Spiegel, Ber. Dtsch. Chem. Ges., 1880, 13, 2, 2219–2221. doi:10.1002/cber.188001302237.
- A. Spiegel, Ber. Dtsch. Chem. Ges., 1881, 14, 1, 873-874. doi:10.1002/cber.188101401183.
- A. Spiegel, Ber. Dtsch. Chem. Ges., 1881, 14, 2, 1686–1696. doi:10.1002/cber.18810140230.
- J. Volhard, Annal. Chem., 1894, 282, 1–21. doi:10.1002/jlac.18942820102.
- L. Claisen, Th. Ewan, Annal. Chem., 1895, 284, 245–299. doi:10.1002/jlac.18952840302.
- R. L. Edwards, M. Gill, J. Chem. Soc., Perkin Trans. 1, 1973, 1921-1929. doi:10.1039/P19730001921.
- N. Ojima, S. Takenaka, S. Seto, Phytochemistry, 1973, 12, 2527–2529.
- N. Ojima, K. Ogura, S. Seto, J. Chem. Soc., Chem. Commun., 1975, 717–718.
- N. Ojima, S. Takenaka, S. Seto, Phytochemistry, 1975, 14, 573-576.
- N. Ojima, I. Takahashi, K. Ogura, S. Seto, Tetrahedron Lett., 1976, 17, 1013–1014.
- I. Takahashi, N. Ojima, K. Ogura, S. Seto, Biochemistry, 1978, 17, 2696–2702.
- M. Kobayashi, N. Ojima, K. Ogura, S. Seto, Chem. Lett., 1979, 579–582.
- H. Sugiyama, N. Ojima, M. Kobayashi, Y. Senda, J. Ishiyama, S. Seto, Agric. Biol. Chem., 1979, 43, 403–404.
- B. Steffan, W. Steglich, Angew. Chem., Int. Ed., 1984, 23, 6, 445–447. doi:10.1002/anie.198404451.
- M. Gill, D. A. Lally, Phytochemistry, 1985, 24, 1351–1354. doi:10.1016/S0031-9422(00)81131-0.
- T. Le Gall, C. Mioskowski, B. Amekraz et al. Angew. Chem., Int. Ed., 2003, 42, 11, 1289–1293. doi:10.1002/anie.198404451.
- D. Klostermeyer, L. Knops, T. Sindlinger, K. Polborn, W. Steglich Eur. J. Org. Chem., 2000, 4, 603–609. doi:10.1002/anie.200390332.
- M. Gill, W. Steglich, Prog. Chem. Org. Nat. Prod., 1987, 51, 1-317. Springer-Verlag.
- K. Rehse, J. Wagenknecht, N. Rietbrock, Arch. Pharm., 1978, 311, 986–991.
- K. Rehse, U. Emisch, Arch. Pharm., 1982, 315, 1020–1025.
- K. Rehse, J. Schinke, G. Bochert, Arch. Pharm., 1979, 312, 390–394.
- K. Rehse, J. Lehmke, Arch. Pharm., 1985, 318, 11–14.
- A. C. Campbell, M. S. Maidment, J. H. Pick, D. F. M. Stevenson, J. Chem. Soc., Perkin Trans. 1, 1985, 1567–1576. doi:10.1039/P19850001567.
- C. E. Caufield, S. A. Antane, K. M. Morris, S. M. Naughton, D. A. Quagliato, P. M. Andrae, A. Enos, J. F. Chiarello, J. (Wyeth, and Brother Ltd., USA), WO 2005019196, US 2005054721, 2005.
- S. A. Antane, C. E. Caufield, W. Hu, D. Keeney, P. Labthavikul, K. M. Morris, S. M. Naughton, P. J. Petersen, B. A. Rasmussen, G. Singh, Y. Yang, Bioorg. Med. Chem. Lett., 2006, 176–180. doi:10.1016/j.bmcl.2005.09.021.
- D. W. Knight, G. Pattenden, J. Chem. Soc., Chem. Commun., 1975, 876–877. doi:10.1039/C39750000876.
- D. W. Knight, G. Pattenden, J. Chem. Soc., Perkin Trans. 1, 1979, 70–76. doi:10.1039/P19790000070.
- P. J. Jerris, P. M. Wovkulich, A. B. Smith III, Tetrahedron Lett., 1979, 20, 4517–4520. doi:10.1016/S0040-4039(01)86637-5.
- M. Gill, M. J. Kiefel, D. A. Lally, A. Ten, Aust. J. Chem. 1990, 43, 1497–1518.
- N. Kaczybura, R. Brückner, Synthesis, 2007, 118–130. doi:10.1055/s-2006-950378.
- D. Bernier, F. Moser, R. Brückner, Synthesis, 2007, 15, 2240–2248. doi:10.1055/s-2007-983800.
- D. Bernier, R. Brückner, Synthesis, 2007, 15, 2249–2272. doi:10.1055/s-2007-983803.