Noonan-Syndrom
Das Noonan-Syndrom ist eine Erbkrankheit. Das komplexe Syndrom ist durch eine Vielzahl von genetischen Entwicklungsstörungen gekennzeichnet. Die resultierenden Fehlbildungen können das äußere Erscheinungsbild betreffen, aber auch innere Organe. So sind angeborene Herzfehler typisch. Da die feststellbaren Symptome denen des Ullrich-Turner-Syndroms sehr ähnlich sind, bezeichnet man das Noonan-Syndrom auch als „Pseudo-Turner-Syndrom“ oder „Male-Turner-Syndrom“. Im Gegensatz zum Ullrich-Turner-Syndrom sind jedoch keine Besonderheiten der Chromosomen (in Bezug auf Anzahl und Struktur) nachweisbar.
| Klassifikation nach ICD-10 | |
|---|---|
| Q87.1 | Angeborene Fehlbildungssyndrome, die vorwiegend mit Kleinwuchs einhergehen
|
| ICD-10 online (WHO-Version 2019) | |
Die Bezeichnung „Noonan-Syndrom“ geht auf die US-amerikanische Kinderkardiologin Jacqueline Noonan zurück, die das Syndrom erstmals 1963[1] und ausführlich 1968 beschrieben hat.[2]
Ursachen
Bei der Hälfte der Menschen mit einem Noonan-Syndrom liegen Veränderungen (Mutationen) des PTPN-11-Gens vor, das sich auf dem langen Arm von Chromosom 12 befindet. Dieses Gen ist verantwortlich für ein Protein (SHP-2), das eine zentrale Funktion in der Wachstumsregulation ausübt, und beeinflusst eine Vielzahl von Stoffwechsel- und Wachstumsvorgängen. Viele dieser Vorgänge werden über die Aktivierung der mitogen-activated protein kinase-Kaskade (MAP-Kinase-Weg) reguliert. Weitere betroffene Gene sind unter anderem das SOS1- und das KRAS-Gen.[3] Inzwischen sind auch in der Proteinkinase RAF1, die am oberen Ende des MAP-Kinase-Wegs steht, Mutationen in Noonan-Patienten gefunden worden.
Es ist zu erwarten, dass für die verbleibenden Fälle weitere verantwortliche Gene im MAP-Kinase-Weg identifiziert werden.
Vererbung
Die Fälle sind zu ca. 50 % sporadisch, d. h., dass Eltern betroffener Kinder selber nicht Träger des Noonan-Syndroms sind. Bei den übrigen Menschen wird das Noonan-Syndrom vererbt, wobei der Erbgang der Genmutation autosomal-dominant verläuft (Autosomen = alle Chromosomen außer den Geschlechtschromosomen). Dies bedeutet, dass bei einem betroffenen Elternteil die Auftrittswahrscheinlichkeit für Kinder bei 50 % liegt (beide Eltern à 75 %).
Die Übertragung geschieht in der Regel durch die Mutter (3:1), da männliche Personen mit dem Noonan-Syndrom meistens unfruchtbar sind.
Häufigkeit
Das Noonan-Syndrom ist relativ häufig. Angaben über die Häufigkeit reichen von 1:500 bis 1:2500. In Deutschland sind ca. 80.000 Personen betroffen. Das Syndrom betrifft beide Geschlechter gleich häufig und ist nach dem Down-Syndrom (Trisomie 21) die zweithäufigste genetische Ursache, die gehäuft zum Auftreten eines Herzfehlers führt.
Diagnose und Symptome
Die vielgestaltigen Ausprägungen, die das Noonan-Syndrom annehmen kann, erschweren die klinische Diagnose. Viele der milderen Fälle werden vermutlich nie diagnostiziert.
Die molekulargenetische Befundung des PTPN-11-Gens kann heute zur Sicherung der Verdachtsdiagnose herangezogen werden. Allerdings haben nur etwa 50 % die gesuchte Mutation. Vor allem lässt die Analyse des Gens bei den Eltern auch eine bessere Einschätzung des Risikos für Folgeschwangerschaften zu. Auch eine vorgeburtliche Diagnose ist bei konkretem Verdacht im Rahmen von Pränataldiagnostik z. B. durch Amniozentese möglich.
Abzugrenzen sind ein Kardio-fazio-kutanes Syndrom, der Cherubismus sowie das Costello-Syndrom.
Symptomatik
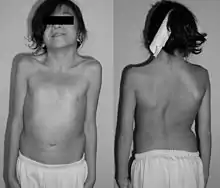
Durch die Fehlinformationen auf dem betroffenen Gen werden verschiedene Symptome und Erkrankungen ausgelöst. Nicht alle Merkmale treten bei allen Kindern auf und in gleicher Ausprägung und mit steigendem Alter werden die Symptome zunehmend unauffälliger. Das macht eine eindeutige Diagnose noch schwieriger.
Viele Symptome erinnern, wie bereits erwähnt, an das Ullrich-Turner-Syndrom. Dazu gehören in erster Linie Kleinwuchs, der beim Noonan-Syndrom allerdings weniger stark ausgeprägt ist, der tiefe Haaransatz im Nacken, sowie gelegentlich das Pterygium colli oder „Flügelfell“. Darunter versteht man eine verbreiterte Nackenhaut mit zur Schulter ziehender Falte.
Die häufigsten möglichen Symptome seien im Folgenden stichwortartig zusammengefasst:
- Wachstumsverzögerungen, Kleinwuchs (bei 40 %) / Durchschnittsgröße bei der Geburt: ca. 47 cm
- Weit auseinander liegende Augen (Hypertelorismus) (bei 95 %)
- Hängende Augenlider (Ptosis)
- geschrägte Lidachsen
- zusätzliche Hautfalte in den inneren Lidwinkeln (Epikanthus medialis)
- Unübliche Hornhautkrümmung (Keratokonus)
- Schielen (Strabismus)
- Breite Nasenflügel
- Ausgeprägtes Philtrum / Einbuchtung in der Mitte der Oberlippe (bei 95 %)
- Hoher Gaumen,
- Kleine Kiefer (Mikrogenie, Mikrognathie),
- Zahnfehlstellungen
- Nach hinten gekippte, tief ansetzende Ohren (bei 90 %)
- Auffallend modellierte Ohrmuscheln („Krempe“)
- Gelegentlich Schwerhörigkeit
- Tiefer Haaransatz (häufig lockiges Haar)
- Pterygium colli („Flügelfell“, s. o.)
- Vertiefung oder Wölbung des Brustbeins („Trichterbrust“)
- weit auseinander liegende Brustwarzen (Mamillen)
- Gelegentlich Skoliose (S-förmig gebogene Wirbelsäule)
- Vergrößerter Haltungswinkel der Arme (Cubitus valgus)
- Vierfingerfurche
- Bei Jungen Unterentwicklung der äußeren Geschlechtsorgane
- Fehlender Eintritt der Hoden in den Hodensack (Kryptorchismus)
- Herzfehler (bei 80 %), z. B. Verengung der Lungenschlagader (Pulmonalstenose), Verdickung des Herzmuskels (hypertrophe Kardiomyopathie)
Es besteht eine Assoziation mit der Juvenilen myelomonozytären Leukämie.[4]
Therapie
Es ist lediglich eine symptomatische Therapie des Noonan-Syndroms möglich, z. B. eine operative Korrektur von Herzfehlern oder des Pterygiums, Tragen von Brillen oder Hörgeräten, Behandlung von Sprachfehlern durch Logopädie oder eine Hormontherapie gegen die Kleinwüchsigkeit. Eine ursächliche Behandlung zur Heilung gibt es zurzeit nicht.
Lernschwierigkeiten und kognitive Einschränkungen
Während die Mehrzahl der Personen mit Noonan-Syndrom einen durchschnittlichen IQ haben, haben knapp 25 % eine leichte kognitive Behinderung (IQ zwischen 69 und 82)[5]: „Die gewöhnlichste Lernschwierigkeit bei Kindern mit Noonan-Syndrom ist die Schwierigkeit in der mündlichen Argumentation. Kein spezifisches Verhaltensmuster ist identifiziert worden, obwohl betroffene Personen häufig Zeichen von Ungeschicklichkeit, Widerspenstigkeit und Reizbarkeit zeigen.“
Literatur
- G. Neuhäuser, H.-C. Steinhausen (Hrsg.): Geistige Behinderung – Grundlagen, Klinische Syndrome, Behandlung und Rehabilitation. Stuttgart / Berlin / Köln 1999, ISBN 3-17-013879-0.
- S. A. Yatsenko, M. del Valle Torrado, P. H. Fernandes, J. Wiszniewska, M. Gallego, J. Herrera, C. A. Bacino: Molecular characterization of a balanced rearrangement of chromosome 12 in two siblings with Noonan syndrome. In: American Journal of Medical Genetics Part A. 149A, Nr. 12, Dezember 2009, S. 2723–2730, doi:10.1002/ajmg.a.33112, PMID 19938085.
- A. A. Romano, J. E. Allanson, J. Dahlgren, B. D. Gelb, B. Hall, M. E. Pierpont, A. E. Roberts, W. Robinson, C. M. Takemoto, J. A. Noonan: Noonan Syndrome: Clinical Features, Diagnosis, and Management Guidelines. In: Pediatrics. Band 126, Nr. 4, Oktober 2010, S. 746–759, doi:10.1542/peds.2009-3207.
Weblinks
- Noonan-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- V. B. Gerritsen: the matchmaker. In: protein spotlight. SIB
Einzelnachweise
- Jacqueline Anne Noonan, Dorothy A. Ehmke: Associated noncardiac malformations in children with congenital heart disease. Midwest Society for Pediatric Research Cincinnati, Ohio, Oct. 25 and 26, 1962. In: The Journal of Pediatrics. Band 63, Nr. 3, September 1963, S. 468–470, doi:10.1016/S0022-3476(63)80441-2.
- Jacqueline A. Noonan: Hypertelorism With Turner Phenotype. In: American Journal of Diseases of Children. Band 116, Nr. 4, Oktober 1968, S. 373–380, doi:10.1001/archpedi.1968.02100020377005.
- G. Gillessen-Kaesbach, Y. Hellenbroich: Humangenetik. In: Pädiatrie. Springer, Berlin / Heidelberg 2019, ISBN 978-3-662-57294-8, S. 3–22, doi:10.1007/978-3-662-57295-5_1.
- Juvenile Myelomonocytic Leukemia. In: NORD (National Organization for Rare Disorders). Abgerufen am 21. Juli 2020 (englisch).
- Katharina Maidhof-Schmid, Ulrike Wößner, Jörg Klepper: Noonan-Syndrom – XX-Turner-Phänotypus – Pseudo-Turner-Syndrom. Hrsg.: Noonan-Kinder e. V. Aschaffenburg Mai 2011 (PDF).