Lowe-Syndrom
Das Lowe-Syndrom, auch okulo-zerebro-renales Syndrom (OCRL) genannt, ist eine seltene, X-chromosomal vererbte Multisystemerkrankung.
| Klassifikation nach ICD-10 | |
|---|---|
| E72.0 | Störungen des Aminosäuretransportes, inkl. Lowe-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
Das Lowe-Syndrom ist sehr selten. Für die Allgemeinbevölkerung wird die Prävalenz auf 1 : 500 000 geschätzt. Es sind dabei alle Ethnien betroffen.[1] Bedingt durch den X-chromosomalen Erbgang sind fast ausschließlich Jungen von der Erkrankung betroffen. Es sind äußerst seltene Fälle von X-Autosom-Translokationen beschrieben, bei denen auch Mädchen am Lowe-Syndrom erkranken.[2]
Klinisches Bild
Das Lowe-Syndrom ist eine systemische Erkrankung, die sich auf mehrere Organsysteme auswirkt. Die betroffenen Patienten haben eine geistige Behinderung (mentale Retardierung), Muskelhypotonie, eine angeborene Katarakt (grauer Star) und eine selektive proximale Tubulopathie.[1]
Katarakt
Schon bei der Geburt weisen alle Patienten eine dichte Katarakt auf, die sich in der Gebärmutter entwickelt (in utero). Die Katarakt wird durch eine veränderte Migration des embryonalen Epithels hervorgerufen.[3] Etwa die Hälfte der Patienten hat zudem ein Glaukom (grüner Star), zum Teil mit Buphthalmus (Augenvergrößerung). Das Glaukom manifestiert sich meist im ersten Lebensjahr.[1]
Proximale Tubulopathie
Bei der selektiven proximalen Tubulopathie handelt es sich um eine Funktionsstörung im proximalen Tubulus, die der des De-Toni-Fanconi-Syndroms entspricht. Man spricht daher auch von einem sekundären Fanconi-Syndrom. Die proximale Tubulopathie kann von Patient zu Patient sehr unterschiedlich ausgeprägt sein, verschlechtert sich aber mit zunehmendem Alter immer weiter. Zum Zeitpunkt ihrer Geburt sind viele Kinder noch ohne Symptome, die aber während der ersten Lebensmonate auftreten und durch Hydrogencarbonat-, Salz- und Wasserverlust gekennzeichnet sind. Diese Mineralverluste führen bei den Kindern zu einer Gedeihstörung. In der zweiten Lebensdekade entwickeln die meisten Patienten ein chronisches Nierenversagen, das zu einer terminalen Niereninsuffizienz führen kann. Letzteres erfordert eine Nierenersatztherapie.[1]
Die weiteren Symptome entsprechen dem De-Toni-Fanconi-Syndrom: Proteinurie und renale tubuläre Azidose. Der Phosphatverlust über die Nieren führt zu einer renalen Rachitis, zu einer Osteomalazie und zu Spontanfrakturen. Die erhöhte Calcium-Ausscheidung über den Harn (Hypercalciurie) bewirkt eine Nephrokalzinose und Nierensteine. Des Weiteren führt die Funktionsstörung im proximalen Tubulus zu einer Aminoacidurie und zu einer erhöhten Kalium-Ausscheidung, die eine Hypokaliämie auslöst.[1][4]
Nervensystem
Bei der Geburt haben die Kinder eine ernsthafte bis sehr schwere Muskelhypotonie, die zu einer Abwesenheit des tiefen Sehnenreflexes (Muskel-Eigen-Reflex) führen kann. Die Hypotonie kann wiederum zu schweren Problemen der Atemwege im ersten Lebensabschnitt führen. Die motorische Entwicklung ist gestört und die Kinder entwickeln die Fähigkeit des selbstständigen Gehens generell erst nach dem dritten Lebensjahr.
Etwa 10 % der Patienten hat eine leichte geistige Behinderung. In vielen Fällen ist sie jedoch mittelschwer bis schwer mit Intelligenzquotienten unterhalb von 50. Der größte Teil der betroffenen (87 %) zeigt autoaggressives und heteroaggressives Verhalten, Reizbarkeit und Wutanfälle. Auch Zwangsstörungen sind häufig.[5] Etwa 50 % der Patienten über 18 Jahren hat epileptische Anfälle. Bis zu 9 % hat Fieberkrämpfe.[1][6][7]
Genetik
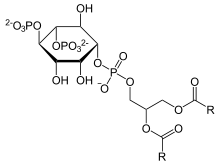
Das Lowe-Syndrom wird durch Mutationen auf dem OCLR1-Gen verursacht. Dieses Gen befindet sich auf dem langen Arm des X-Chromosoms auf Genlocus q25-q26. Das OCLR1-Gen besteht aus 24 Exons und codiert für das Enzym Phosphatidylinositol-4,5-bisphosphat-5-Phosphatase (Inositol-Polyphosphat-5-Phosphatase). Diese Phosphatase gehört zur Familie der 5-Phosphatasen vom Typ II und befindet sich normalerweise im trans-Golgi-Netzwerk.[1] Dort wird sie für den Polymerisationsprozess von Actin benötigt. Es dephosphoryliert dabei insbesondere Phosphatidylinositol-4,5-Bisphosphat [PI(4,5)P2] zu Phosphatidylinositol-4-Phosphat [PI(4)P].[8]
Mutationen in diesem Gen finden sich auf teilweise beim Dent-Syndrom.
Durch den Mangel des Enzyms reichert sich das Substrat Phosphatidylinositol-4,5-bisphosphat in den Zellen der betroffenen Patienten an.
Diagnose
Anhand der Symptome kann eine vorläufige Diagnose gestellt werden. Wegen des ausgesprochen seltenen Vorkommens der Erkrankung ist in jedem Fall eine labormedizinische Absicherung der Diagnose angebracht. Bedingt durch die allelische Heterogenität der Mutationen im OCRL1-Gen, kann eine pränatale DNA-Analyse nur bei Familien durchgeführt werden, bei denen die Mutation bereits bekannt ist. Die Messung der Aktivität der Phosphatidylinositol-4,5-bisphosphat-5-Phosphatase in kultivierten Amniozyten ist ein pränatales biochemisches Verfahren, das zur Diagnose des Lowe-Syndroms verwendet werden kann.[9]
Differentialdiagnostisch ist unter anderem das Dent-Syndrom abzugrenzen.
Therapie
Das Lowe-Syndrom ist unheilbar. Die Therapie erfolgt rein symptomatisch und beinhaltet unter anderem Kataraktoperation, Glaukombehandlung, Sprach- und Physiotherapie. Gegen die Verhaltensstörungen können Neuroleptika, Stimulanzien, Benzodiazepine und Antidepressiva (beispielsweise Serotonin-Wiederaufnahmehemmer) verschrieben werden. Mit Clomipramin, Paroxetin und Risperidon wurden gute Behandlungsergebnisse erzielt.[1]
Prognose
Für die meisten Patienten führt die terminale Niereninsuffizienz oder die Hypotonie zu einem frühzeitigen Tod; typischerweise zwischen dem 30. und 40. Lebensjahr.[1]
Erstbeschreibung
Das okulo-zerebro-renale Syndrom wurde erstmals 1952 von dem US-amerikanischen Pädiater Charles Upton Lowe und Kollegen als Syndrom mit Azidurie, verminderter Harnstoffproduktion, Hydrophthalmus und mentaler Retardierung beschrieben.[10]
Weiterführende Literatur
- J. K. Brooks, R. Ahmad: Oral anomalies associated with the oculocerebrorenal syndrome of Lowe: case report with multiple unerupted teeth and pericoronal radiolucencies. In: Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. Band 107, Nummer 3, März 2009, S. e32–e35, ISSN 1528-395X. doi:10.1016/j.tripleo.2008.11.023. PMID 19217010. (Review).
- A. C. Ruellas, M. M. Pithon u. a.: Lowe syndrome: literature review and case report. In: Journal of orthodontics. Band 35, Nummer 3, September 2008, S. 156–160, ISSN 1465-3125. doi:10.1179/146531207225022599. PMID 18809779. (Review).
- M. T. Rodrigues Santos, M. M. Watanabe u. a.: Oculocerebrorenal Lowe syndrome: a literature review and two case reports. In: Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric Dentistry. Band 27, Nummer 3, 2007 May-Jun, S. 108–111, ISSN 0275-1879. PMID 17658186. (Review).
- M. Lowe: Structure and function of the Lowe syndrome protein OCRL1. In: Traffic. Band 6, Nummer 9, September 2005, S. 711–719, ISSN 1398-9219. doi:10.1111/j.1600-0854.2005.00311.x. PMID 16101675. (Review).
- M. Harrison, E. W. Odell, E. C. Sheehy: Dental findings in Lowe syndrome. In: Pediatric dentistry. Band 21, Nummer 7, 1999 Nov-Dec, S. 425–428, ISSN 0164-1263. PMID 10633515. (Review).
- M. Addis, M. Loi u. a.: OCRL mutation analysis in Italian patients with Lowe syndrome. In: Human mutation. Band 23, Nummer 5, Mai 2004, S. 524–525, ISSN 1098-1004. doi:10.1002/humu.9239. PMID 15108291.
- G. Amirhakimi, M. H. Fallahzadeh, H. Saneifard: Lowe Syndrome: Report of a Case and Brief Literature Review. In: Iran J Pediatr. Band 19, Nummer 4, 2009, S. 417–420.
- T. Menke, S. M. Gu u. a.: Lowe-Syndrom – Klinische Befunde und Nachweis einer Genmutation bei einem 4jährigen Jungen. In: Kinderheilkunde. Band 146, Nummer 2, S. 125–128, doi:10.1007/s001120050257
- Lowe-Syndrom (PDF; 414 kB) Kindernetzwerk e.V.
Einzelnachweise
- M. Loi: Lowe syndrome. In: Orphanet Journal of Rare Diseases. Band 1, 2006, S. 16, ISSN 1750-1172. doi:10.1186/1750-1172-1-16. PMID 16722554. PMC 1526415 (freier Volltext). (Review).
- O. T. Mueller, J. K. Hartsfield u. a.: Lowe oculocerebrorenal syndrome in a female with a balanced X;20 translocation: mapping of the X chromosome breakpoint. In: American Journal of Human Genetics. Band 49, Nummer 4, Oktober 1991, S. 804–810, ISSN 0002-9297. PMID 1897526. PMC 1683175 (freier Volltext).
- R. C. Tripathi, G. W. Cibis, B. J. Tripathi: Pathogenesis of cataracts in patients with Lowe's syndrome. In: Ophthalmology. Band 93, Nummer 8, August 1986, S. 1046–1051, ISSN 0161-6420. PMID 3763153.
- L. R. Charnas, I. Bernardini u. a.: Clinical and laboratory findings in the oculocerebrorenal syndrome of Lowe, with special reference to growth and renal function. In: The New England journal of medicine. Band 324, Nummer 19, Mai 1991, S. 1318–1325, ISSN 0028-4793. doi:10.1056/NEJM199105093241904. PMID 2017228.
- L. Kenworthy, T. Park, L. R. Charnas: Cognitive and behavioral profile of the oculocerebrorenal syndrome of Lowe. In: American journal of medical genetics. Band 46, Nummer 3, Mai 1993, S. 297–303, ISSN 0148-7299. doi:10.1002/ajmg.1320460312. PMID 8488875.
- L. Charnas, J. Bernar u. a.: MRI findings and peripheral neuropathy in Lowe's syndrome. In: Neuropediatrics. Band 19, Nummer 1, Februar 1988, S. 7–9, ISSN 0174-304X. doi:10.1055/s-2008-1052393. PMID 2834662.
- K. McSpadden, Z. Dolinsky, E. Schroerlucke: Report on the Lowe's syndrome comprehensive survey. West Lafayette: Lowe Syndrome Association; 1991.
- V. A. van Rahden: Auswirkungen einer Depletion der humanen Inositol-Polyphosphat 5-Phosphatase OCRL auf Transportwege des Mannose 6-Phosphat-Rezeptors. (PDF; 5,6 MB) Dissertation, Universität Hamburg, 2011, S. 1.
- Lowe-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- C. U. Lowe, M. Terrey, E. A. MacLachlan: Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. In: American Journal of Diseases of Children. Band 83, Nummer 2, Februar 1952, S. 164–184, ISSN 0096-8994. PMID 14884753.
Weblinks
- Lowe-Syndrom. In: Online Mendelian Inheritance in Man. (englisch)
- Lowe-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).