Larsen-Syndrom
Das Larsen-Syndrom, auch Rotter-Erb-Syndrom ist eine hereditäre Bindegewebserkrankung mit multiplen Luxationen und Skelettdysplasie.
| Klassifikation nach ICD-10 | |
|---|---|
| Q68.8 | Sonstige näher bezeichnete angeborene Muskel-Skelett-Deformitäten |
| Q65.2 | Angeborene Luxation des Hüftgelenkes, nicht näher bezeichnet |
| Q68.2 | Angeborene Deformität des Knies |
| ICD-10 online (WHO-Version 2019) | |
Die Erstbeschreibung erfolgte durch Rotter und Erb 1948, die Krankheit ist nach dem US-amerikanischen Orthopäden Loren Joseph Larsen (* 1914) benannt, der 1950 eine Beschreibung veröffentlichte.[1][2]
Verbreitung
Die Häufigkeit wird mit 1–9 zu 1.000.000 angegeben[3], auf der Insel Réunion beträgt die Prävalenz bei Geburt 1 zu 1.500.[4]
Einteilung und Ursache
Je nach Erbgang können unterschieden werden:
- autosomal-dominante Form, die weitaus häufigste. Dieser Erkrankung liegen Mutationen im FLNB-Gen am Genort 3p14.3 zugrunde, welches für Filamin B kodiert.[5][4]
- autosomal-rezessive Form, (Synonyme: CHST3-assoziierte Skelettdysplasie; englisch SPONDYLOEPIPHYSEAL DYSPLASIA, OMANI TYPE), Mutationen im CHST3-Gen an 10q22.1.[6][7]
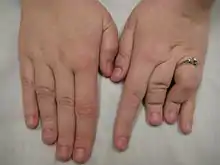
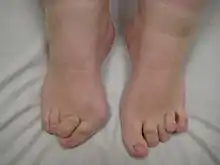
Klinik
Diagnostische Kennzeichen sind:
- Bei Geburt bestehende Luxationen mehrerer Gelenke: Hüften, Knie, Ellenbogen
- Kraniofaziale Anomalie mit eingesunkener Nasenwurzel, Hypertelorismus, flaches Gesicht
- Gaumenspalte, in 50 % Klumpfuß
- Radiologisch breite Endphalangen, Skoliose der oberen Halswirbelsäule, eventuell mit Segmentationsstörung, akzessorischer Knochenkern am Fersenbein, überzählige Handwurzelknochen.
- Atemstörungen.[2][8]
Ferner wurden kardiovaskuläre Störungen beschrieben.[9]
Differentialdiagnose
Klinisch anzugrenzen ist:
- Ehlers-Danlos-Syndrom
- Arthrogryposis multiplex congenita[8]
- Angeborene Knieluxation
- Desbuquois-Syndrom
Differential-diagnostisch sind nach dem Röntgenbefund abzugrenzen:
Diagnostik
Bereits pränatal ist die Verdachtsdiagnose stellbar.[11]
Nach der Geburt ist neben der Dokumentation des Ausmaßes der Luxationen durch Ultraschall zur Abgrenzung der aufgeführten Differentialdiagnosen eine Röntgenaufnahme erforderlich.
Behandlung
Eine kausale Therapie ist nicht möglich. Die Behandlung erfolgt symptombezogen, richtet sich nach dem Ausmaß der Fehlstellungen. Probleme bei der Narkose können durch die Instabilität der Halswirbelsäule auftreten.[9]
Siehe auch
Einzelnachweise
- N. Mitra, N. Kannan, V. S. Kumar, G. Kavita: Larsen Syndrome: A Case Report. In: Journal Of Nepal Paediatric Society. 32.1 (2012), S. 85–87.
- G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger (Hrsg.); B. Leiber: Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. 7. Auflage. Urban & Schwarzenberg, 1990, ISBN 3-541-01727-9.
- M. Vujic, K. Hallstensson, J. Wahlström, A. Lundberg, C. Langmaack, T. Martinson: Localization of a gene for autosomal dominant Larsen syndrome to chromosome region 3p21.1-14.1 in the proximity of, but distinct from, the COL7A1 locus. In: American Journal of Human Genetics. Band 57, Nummer 5, November 1995, S. 1104–1113, ISSN 0002-9297. PMID 7485161. PMC 1801374 (freier Volltext).
- Larsen-Syndrom, autosomal-dominates. In: Orphanet (Datenbank für seltene Krankheiten).
- Larsen syndrome . In: Online Mendelian Inheritance in Man. (englisch)
- CHST3-assoziierte Skelettdysplasie. In: Orphanet (Datenbank für seltene Krankheiten).
- Spondyloepiphyseal dysplasia with congenital joint dislocations. In: Online Mendelian Inheritance in Man. (englisch)
- F. Hefti: Kinderorthopädie in der Praxis. Springer 1998, ISBN 3-540-61480-X, S. 662.
- E. A. Kiel, J. L. Frias, B. E. Victorica: Cardiovascular manifestations in the Larsen syndrome. In: Pediatrics. Band 71, Nummer 6, Juni 1983, S. 942–946, ISSN 0031-4005. PMID 6856406.
- J. W. Spranger: Bone Dysplasias. Urban & Fischer 2002, ISBN 3-437-21430-6, S. 241.
- T. Tongsong, C. Wanapirak, S. Pongsatha, J. Sudasana: Prenatal sonographic diagnosis of Larsen syndrome. In: Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in Medicine. Band 19, Nummer 6, Juni 2000, S. 419–421, ISSN 0278-4297. PMID 10841065.