Holoprosencephalie
Die Holoprosencephalie (HPE) ist eine pränatal (vorgeburtlich) entstandene Fehlbildung im Bereich des Vorderhirns und des Gesichts.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| Q04.2 | Holoprosenzephalie-Syndrom |
| ICD-10 online (WHO-Version 2019) | |
Häufigkeit
In etwa einer bis vier von 1.000 Schwangerschaften kommt Holoprosencephalie beim Kind vor und stellt somit die häufigste angeborene Gehirnfehlbildung dar. Da jedoch die meisten Babys nicht lebensfähig sind, wird aufgrund der hohen intrauterinen Letalität nur eines von 2.500 bis 16.000 Kindern mit Holoprosencephalie lebend geboren. Mädchen sind häufiger betroffen als Jungen (Gynäkotropie) und die Fehlbildung wird überdurchschnittlich häufig bei Kindern sehr junger Mütter festgestellt.[3][1]
Entstehung
Die Fehlbildung entsteht in der etwa dritten bis sechsten Embryonalwoche durch eine unvollständige Teilung des Vorderhirns infolge einer Störung im Bereich des Mittellinien-Entwicklungsfeldes des Kopfes: Es kommt nicht zur vollständigen Teilung bzw. Differenzierung des Prosencephalons (Vorderhirn), das sich aus dem Telencephalon (Endhirn) und dem Diencephalon (Zwischenhirn) zusammensetzt.[3]
Ursachen
Zu den genauen Ursachen (Ätiologie) der Holoprosencephalie ist noch nicht viel bekannt. In den meisten Fällen ist das Auftreten zufällig (sporadisch), aber offenbar kann ein genetisch bedingter Mangel an Cholesterin die Entwicklungsstörung (mit)verursachen. Als Risikofaktoren gelten zudem Diabetes mellitus der Schwangeren, Viruserkrankung des Ungeborenen, Toxoplasmose, verschiedene Teratogene und Umweltfaktoren wie z. B. Hyperglykämie, Hypocholesterinämie, Retinolsäure und Ethanol, die Einfluss nehmen können.
Statistisch gesehen finden sich bei rund 50 von 100 betroffenen Kindern klare Abweichungen auf chromosomaler Ebene (z. B. Trisomien) und bei weiteren 20 können durch spezielle molekulargenetische Techniken Veränderungen erkannt werden. Die Ursache der Holoprosencephalie bei den übrigen 30 % der Kinder kann derzeit nicht mit genetischen Gründen in Zusammenhang gebracht werden.
Mehr als 25 Erkrankungen mit genetischem Hintergrund sind mit einer erhöhten Wahrscheinlichkeit für eine Holoprosencephalie assoziiert. Dazu zählen auch Chromosomenbesonderheiten wie Triploidie, Trisomie 13 (Pätau-Syndrom), Trisomie 18 (Edwards-Syndrom), 18p-Syndrom, Pseudotrisomie-13-Syndrom, Chromosom-7q-Syndrom, De-Grouchy-Syndrom, Kurzripp-Polydaktylie-Syndrome und das Joubert-Syndrom.[4]
In Untersuchungen zu genetischen Faktoren konnten bislang verschiedene Holoprosencephalie-Loci (HPE) gefunden werden:
- 4 häufige SHH auf 7q36, ZIC2 auf 13q32, SIX3 auf 2p21, TGIF auf 18p11
- 10 seltene PTCH1 auf 9q22, GLI2 auf 2q14, FOXH1 auf 8q24, TDGF1 auf 3p21, DISP1 auf 1q42, NODAL auf 10q22, FGF8 auf 10q24, GAS1 auf 9q21, DLL1 auf 6q27 und CDON auf 11q23-q24
Der Erbgang scheint autosomal-dominant oder autosomal-rezessiv zu sein.[1][5]
Im Rahmen von Syndromen
- Agnathie - Holoprosenzephalie - Situs inversus[6]
- Hartsfield-Syndrom, Synonym: Holoprosenzephalie-Ektrodaktylie-Lippen-Kiefer-Gaumenspalte-Syndrom[7]
- Genoa-Syndrom, Synonyme: Camera-Lituania-Cohen-Syndrom; Holoprosenzephalie - Kraniosynostose[8]
- Pseudotrisomie-13-Syndrom, Synonym: Holoprosenzephalie - postaxiale Polydaktylie
- Morse-Rawnsley-Sargent-Syndrom, Synonyme: Holoprosenzephalie-fetale Akinesie/Hypokinesie-Sequenz-Syndrom; Holoprosenzephalie-Hypokinesie-kongenitale Kontrakturen-Syndrom[9]
- Steinfeld-Syndrom, Synonym: Holoprosenzephalie - Radius-, Herz- und Nierenfehlbildungen[10]
- Stenose der Apertura piriformis, kongenitale, mit Holoprosenzephalie[11]
Klinische Merkmale
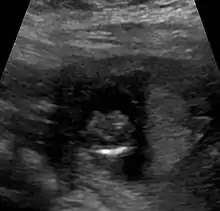
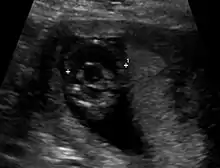
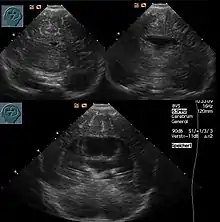
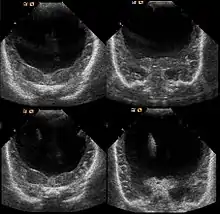
Die Ausprägung weist ein breites Spektrum auf: klinisch unauffällige Mutationsträgerschaft, Lippen-Kiefer-Gaumen-Spalten (auch mediane Pseudospalten), Mikroglossie, leichte Formen (z. B. mit nur einem zentralen Schneidezahn, Hypotelorismus / nah beieinander liegenden Augen) kommen ebenso vor wie Arrhinenzephalie, Corpus-callosum-Agenesie, Agenesie der Hypophyse sowie Störungen mit Zyklopie. Etwa neun von zehn Kindern haben weitere Fehlbildungen.
Einteilung
Die klassische Einteilung nach zunehmendem Schweregrad und anatomischen Veränderungen nach DeMyer unterscheidet:[12]
- lobäre Holoprosencephalie (Trennung ist größtenteils erfolgt, kompletter Interhemisphärenspalt, zwei Seitenventrikel mit rudimentärer Verbindung)[13]
- semilobäre Holoprosencephalie (teilweise Trennung, hinterer Interhemisphärenspalt mit rudimentären Hemisphären, ein Hirnventrikel)[14]
- alobäre Holoprosencephalie (keine Trennung, kein Interhemisphärenspalt, ein Hirnventrikel)[15]
Hinzu kommen Mikroformen
- Mikroforme Holoprosencephalie[16]
- Syndrom des einzelnen maxillären mittleren Schneidezahnes
und Sonderformen
Diagnose
Die Diagnose kann vorgeburtlich im Rahmen von Pränataldiagnostik durch insbesondere Feinultraschalluntersuchungen im zweiten Trimenon, teils aber auch schon früher, gestellt werden. Während die Feststellung der alobären und der semilobären Form oft recht einfach ist, ist die der lobären Holoprosencephalie komplizierter.
Nach gesicherter vorgeburtlicher Diagnose können sich die werdenden Eltern zu einem Schwangerschaftsabbruch aus medizinischer Indikation entscheiden oder sich durch das Wissen auf die Geburt des Kindes einstellen und entsprechende Vorbereitungen treffen (Klinikwahl usw.).
Nachgeburtlich sind die Schnittbildverfahren Sonographie und Magnetresonanztomographie Methoden der Wahl.[19]
Differentialdiagnose
Abzugrenzen sind:[1]
- Anenzephalie
- angeborener Hydrozephalus erheblicher Ausprägung
- Walker-Warburg-Syndrom
- große Zyste im Interhemisphärenspalt
- Otozephalie
- andere Mittelliniendefekte
Therapie und Folgen
Derzeit ist keine Therapie bekannt, lediglich die Symptome können gegebenenfalls behandelt werden (symptomatische Therapie). Die Sterberate bei betroffenen Kindern ist in der Schwangerschaft sehr hoch. Die Prognose zu Lebenserwartung und Entwicklung von neugeborenen Kindern mit Holoprosencephalie ist meist ungünstig: Kinder mit einer schweren Form der Fehlbildung versterben meist innerhalb der ersten Monate nach der Geburt.
Die Prognose ist bei der alobaren Form schlechter als bei der semilobaren oder lobaren Form. Ein Erfahrungswert ist, dass ca. 9 von 10 Kindern mit der alobaren Form innerhalb des ersten Lebensjahres versterben. Kinder, die das erste Jahr überleben, können das Erwachsenenalter durchaus erreichen. Bei ihnen sind je nach Schweregrad kognitive und körperliche Beeinträchtigungen zu erwarten sowie neurologische Auffälligkeiten (z. B. Epilepsie), fehlende oder eingeschränkte Lautsprachentwicklung, Schlafschwierigkeiten u. a.
Literatur
- C. Dubourg, C. Bendavid, L. Pasquier, C. Henry, S. Odent, V. David: Holoprosencephaly. In: Orphanet J Rare Dis. 2;2, Feb 2007, S. 8. PMID 17274816, PMC 1802747 (freier Volltext)
- P. Reimer, P. M. Parizel, F.-A. Stichnoth (Herausgeber): Clinical MR Imaging. A Practical Approach. Springer, 2. Auflage 2006, ISBN 3-540-31530-6
Einzelnachweise
- Holoprosenzephalie. In: Orphanet (Datenbank für seltene Krankheiten).
- W. Schuster, D. Färber (Herausgeber): Kinderradiologie. Bildgebende Diagnostik. Springer 1996, ISBN 3-540-60224-0. Bd. 1, S. 482ff
- GeneReviews
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- Emedicine
- Agnathie - Holoprosenzephalie - Situs inversus. In: Orphanet (Datenbank für seltene Krankheiten).
- Hartsfield-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie - Kraniosynostose. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie-Hypokinesie-kongenitale Kontrakturen-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Steinfeld-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- Stenose der Apertura piriformis, kongenitale, mit Holoprosenzephalie. In: Orphanet (Datenbank für seltene Krankheiten).
- W. DeMyer, W. Zeman, C. G. Palmer: The Face Predicts The Brain: Diagnostic Significance Of Median Facial Anomalies For Holoprosencephaly (Arhinencephaly). In: Pediatrics. Band 34, August 1964, S. 256–263, PMID 14211086.
- Holoprosenzephalie, lobäre. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie, semilobare. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie, alobäre. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie, mikroforme. In: Orphanet (Datenbank für seltene Krankheiten).
- Mittlere interhemisphärische Fusionsvariante der Holoprosenzephalie. In: Orphanet (Datenbank für seltene Krankheiten).
- Holoprosenzephalie, septopräoptische. In: Orphanet (Datenbank für seltene Krankheiten).
- V. Hofmann, K. H. Deeg, P. F. Hoyer: Ultraschalldiagnostik in Pädiatrie und Kinderchirurgie. Lehrbuch und Atlas. Thieme 2005, ISBN 3-13-100953-5
{kind=link}
{kind=link}