Faktor-V-Leiden-Mutation
Die Faktor-V-Mutation Typ Leiden („Faktor-fünf-Mutation Typ Leiden“; häufig als FVL-Mutation abgekürzt) ist der häufigste angeborene thrombophile Risikomarker. Das Thromboserisiko ist bei heterozygoten Anlageträgern um das 4- bis 7-fache gegenüber der Normalbevölkerung erhöht. Bei homozygoten Anlageträgern dürfte das Thromboserisiko um das etwa 11,5- bis 26-fache erhöht sein.[1][2] „Leiden“ steht für die niederländische Universitätsstadt, wo die Mutation 1994 zuerst beschrieben wurde.[3] Diese genetische Variante führt zu einer Resistenz gegen aktiviertes Protein C, d. h. Faktor V kann durch aktiviertes Protein C nur unzureichend inaktiviert werden. Das veränderte Basentriplett führt zum Einbau der Aminosäure Glutamin an Stelle von Arginin an Position 506 des Proteins. Der so entstandene Gerinnungsfaktor wird Faktor-V Leiden, kurz FVL, genannt. Durch die Veränderung in der Proteinsequenz kommt die so genannte APC-Resistenz zustande: Normalerweise wird der aktivierte Faktor V (FVa) durch das aktivierte Protein C (APC) durch Proteolyse abgebaut und damit wirkungslos gemacht. Durch die veränderte Struktur im FVL wird der Abbau von Faktor Va durch APC inhibiert (er wird „resistent“), und der Faktor Va behält seine gerinnungsfördernde Wirkung. Hierdurch kommt es zu einem Ungleichgewicht an gerinnungshemmenden und gerinnungsfördernden Einflüssen, wodurch die Neigung, Thrombosen zu entwickeln, zunimmt (Thrombophilie).
Epidemiologie
In Europa sind etwa 5 % der Bevölkerung heterozygote Träger der FVL-Mutation. Nur 0,05–0,5 % sind homozygote Träger, die je ein mutiertes Allel von Vater und Mutter geerbt haben (sofern es keine Neumutation ist). Bei Menschen nichteuropäischer Abstammung kommt die Mutation noch deutlich seltener vor.[2] Evolutionsbiologisch bot die Mutation in früheren Zeiten bei größeren Verletzungen einen Vorteil, da sie die Wahrscheinlichkeit des Verblutens verringerte, dies ist vermutlich für ihre heutige Häufigkeit verantwortlich. Durch natürliche Selektion, Gründereffekte und Gendrift kann die Mutation in einigen kleineren Populationen auch noch deutlich gehäufter auftreten, so etwa bei 12 % der Schweden und Zyprer.[4]
Geschichtliche Aspekte
Ein Blutgerinnungsfaktor-V-Mangel wurde 1955 erstmals von Max-Hermann Hörder entdeckt und auf einen Blutgerinnungsfaktor-V-Inhibitor (FVI) zurückgeführt. Im Jahre 1993 wurde dann zum ersten Mal durch den schwedischen Arzt Björn Dahlbäck die Faktor-V-Leiden-Mutation (FVL-Mutation) beschrieben, welche letztlich eine verstärkte Blutgerinnungsneigung bewirkt. Dahlbäck hatte bereits 1989 bei einem jungen Mann eine ungewöhnliche Häufung von Venenthrombosen beobachtet, auch bei anderen Familienmitgliedern des Mannes waren bereits Thrombosen aufgetreten. Für eine genaue Untersuchung von Blutproben der Familie mussten zunächst Untersuchungsmethoden neu entwickelt und verfeinert werden. Schließlich gelang der Nachweis einer Punktmutation in dem für Gerinnungsfaktor V codierenden Gen, das auf dem langen Arm von Chromosom 1 (Genlocus 1q24.2) liegt. Durch die Mutation wird ein einzelnes Nukleotid an Position 1691 verändert (Adenin statt Guanin). Dahlbäck benannte diese genetische Veränderung, wie es unter Genomforschern üblich ist, nach dem Ort ihrer Entdeckung – der niederländischen Stadt Leiden – als Faktor-V-Leiden-Mutation.
Diagnostik der FVL-Mutation
Indikation zur Untersuchung
Personen, in deren engerem Verwandtschaftskreis – hierzu gehören Großeltern, Eltern, Geschwister und eigene Kinder – bereits mehrfach ungeklärte Thrombosen aufgetreten sind, können sich einer Gerinnungsdiagnostik unterziehen. Hierzu gehört unter anderem auch die Untersuchung auf die FVL-Mutation bzw. die APC-Resistenz. Wenn bei einer Person selbst eine oder mehrere thromboembolische Erkrankungen auftraten, wird ebenfalls eine Gerinnungsdiagnostik empfohlen. Dies gilt umso mehr, wenn für das Auftreten der Thrombose keine etablierten Risikofaktoren (Übergewicht, Immobilisierung, z. B. nach Operationen oder Frakturen, Einnahme von hormonellen Kontrazeptiva etc.) vorliegen. In den letzten Jahren wurde bei Patientinnen mit wiederholten Fehlgeburten (sog. habituellen Aborten), Totgeburten ansonsten unklarer Ursache sowie bei schwerer intrauteriner Wachstumsretardierung ebenfalls ein Zusammenhang mit einer mütterlichen Thromboseneigung (Thrombophilie) festgestellt,[5] so dass auch in diesen Fällen eine Gerinnungsuntersuchung der Mutter und gegebenenfalls eine Prophylaxe angebracht ist.
Verfahren
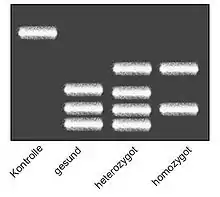
Die Punktmutation im Faktor-V-Gen von Guanin (G) zu Adenin (A) an Position 1691 lässt sich durch DNA-Sequenzierung nachweisen.[6] Da die Methode zum Direktnachweis jedoch sehr teuer ist, wird die Mutation meist über einen Restriktionsfragmentlängenpolymorphismus (RFLP) nachgewiesen.
Dazu wird die DNA einer Blutprobe des Patienten mit einer Polymerase-Kettenreaktion (PCR) vervielfältigt und das betreffende Gen durch eine chemische Reaktion mithilfe eines Restriktionsenzymes (MnlI) in verschieden lange Nukleinsäureketten zerschnitten. Die Länge der Nukleinsäureketten wird anschließend in einer Gelelektrophorese bestimmt. Restriktionsfragmente von PCR-Produkten, welche die FVL-Mutation tragen, weisen eine andere Größenverteilung auf als solche, die von gesunden Probanden stammt,[7] da durch die Mutation eine Erkennungsstelle für das Restriktionsenzym entfernt wird.
Der aktuelle Standard ist die Bestimmung über die Messung der Schmelzkurve. Hierbei bedient man sich des Verfahrens der "Real-Time-PCR". Es wird eine Polymerasekettenreaktion mit fluoreszenzmarkierten Nukleinsäuresonden durchgeführt, welche zu der ggf. von der Mutation betroffenen Sequenz komplementär sind. Der primäre Farbstoff an diesen Sonden wird mit einer bestimmten Wellenlänge angeregt und emittiert Licht einer Wellenlänge, mit der ein sekundärer Fluoreszenzfarbstoff angeregt wird, der an eine zweite Sonde gekoppelt ist, die zu einer benachbarten Gensequenz komplementär ist. Nur wenn beide Sonden tatsächlich an die DNA gebunden sind, sind sie räumlich nah genug für diese Kooperation (FRET genannt). So lässt sich im Rahmen eines Temperaturgradienten photometrisch die Schmelztemperatur des Gen-Sonden-Doppelstrangs bestimmen. Wenn die Mutation vorliegt, binden die Sonden nicht vollständig komplementär, sodass die Schmelztemperatur erniedrigt ist. Bei einem heterozygoten Genotypen entsteht entsprechend eine „doppelte“ Schmelzkurve.[8]
In jüngerer Zeit wird die Mutation zunehmend auch zufällig im Rahmen der personalisierten Medizin und ihren genetischen Mikrochip-Untersuchungen entdeckt, der maßgebliche Einzel-Nukleotid-Polymorphismus (SNP) trägt den Namen rs6025.[2]
Vererbung der FVL-Mutation
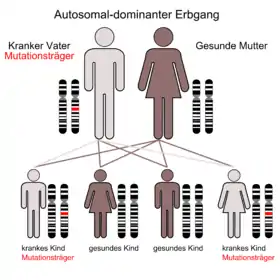
Die FVL-Mutation wird autosomal dominant vererbt. Dies bedeutet, dass auch Personen, welche die FVL-Mutation von nur einem Elternteil erben (heterozygot), bereits ein 5- bis 10-fach erhöhtes Risiko haben, eine Thrombose zu erleiden. Allein 8 % der Bevölkerung Bayerns weisen den thrombophilen Risikomarker (kein Erbfehler) in dieser Form auf. Wenn beide Elternteile die FVL-Mutation an ihr Kind weitergeben (homozygot), so besteht vermutlich ein 26-fach erhöhtes Thromboserisiko (maximal 7x7=49).
Leben mit der Faktor-V-Leiden-Mutation
Anlageträger der Faktor-V-Leiden-Mutation sollten einen thrombosefördernden Lebensstil meiden, um die Wahrscheinlichkeit einer Thrombose zu verringern. Hierzu zählen insbesondere der Verzicht auf das Rauchen, das Vermeiden von Übergewicht sowie, für junge Frauen, das Absehen von der Einnahme östrogenhaltiger oraler Kontrazeptiva. Langes Sitzen, etwa auf Flug- oder Busreisen, sollte häufiger durch Bewegungen der Beine unterbrochen werden. Solange Thrombosen vermieden werden, ist die Lebenserwartung für gewöhnlich nicht beeinträchtigt und bei ansonsten gesunden Heterozygoten keine Gabe von gerinnungshemmenden Medikamenten erforderlich. Homozygote Anlageträger hingegen können aufgrund des sehr hohen Thromboserisikos unter Umständen auf eine lebenslange Therapie angewiesen sein, insbesondere wenn weitere Risikomarker vorliegen.
In der Schwangerschaft kann jedoch auch bei heterozygoten Frauen zur Vorbeugung von Spontanaborten und Blutgerinnseln eine Thromboseprophylaxe mit niedermolekularem Heparin angeraten sein, ebenso nach größeren Operationen mit Bettlägerigkeit, wo insbesondere eine rasche Mobilisation von großer Wichtigkeit ist.
Literatur
- Bjorn Dahlbäck: The discovery of activated protein c resistance. In: Journal of Thrombosis and Haemostasis. Band 1, Nr. 1, 2003, S. 3–9.
- David H. Lee et al.: Prevalence of factor V Leiden in a Canadian blood donor population. In: Canadian Medical Association Journal. Band 155, 1996, S. 285 (lac-bac.gc.ca).
- Thrombophilia as a multigenic disease. In: Haematologica. Band 84, Nr. 1, 1999, S. 59–70.
- Evelyn Rey et al.: Thrombophilic disorders and fetal loss: a meta-analysis. In: The Lancet. Band 361, Nr. 9361, 15. März 2003, S. 901–908, PMID 12648968.
Einzelnachweise und Anmerkungen
- Zotz, Sucker, Gerhardt: Bedeutung thrombophiler Risikofaktoren für das Erst- und Rezidivthromboserisiko. (PDF) In: DRK – hämotherapie. Abgerufen am 9. September 2018.
- Artikel zu rs6025 auf snpedia.com, abgerufen am 9. Januar 2019.
- Rogier M. Bertina, Bobby P. C. Koeleman, Ted Koster, Frits R. Rosendaal, Richard J. Dirven: Mutation in blood coagulation factor V associated with resistance to activated protein C. In: Nature. Band 369, Nr. 6475, Mai 1994, ISSN 0028-0836, S. 64–67, doi:10.1038/369064a0 (nature.com [abgerufen am 9. September 2018]).
- Zeitbombe im Blut. In: Der Spiegel. Nr. 15, 1997, S. 222 f. (online – 7. April 1997).
- Rey et al., 2003
- siehe Bild
- siehe Bild, Spalten 2, 3 und 4
- S H Neoh, M J Brisco, F A Firgaira, K J Trainor, D R Turner: Rapid detection of the factor V Leiden (1691 G > A) and haemochromatosis (845 G > A) mutation by fluorescence resonance energy transfer (FRET) and real time PCR. In: Journal of Clinical Pathology. Band 52, Nr. 10, Oktober 1999, ISSN 0021-9746, S. 766–769, PMID 10674036, PMC 501573 (freier Volltext).