Cystic Fibrosis Transmembrane Conductance Regulator
Der Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) ist ein auf der Oberfläche von Zellen festsitzendes Protein, ein sogenannter Chloridkanal, der hauptsächlich in der Zellmembran von Epithelzellen von Fischen und Landwirbeltieren vorkommt. Mutationen im CFTR-Gen beim Menschen führen zum Fehlen oder zur eingeschränkten Funktion des Kanals, was Ursache der Mukoviszidose (zystischen Fibrose) und der kongenitalen Aplasie des Vas deferens (CAVD) ist.
| Cystic Fibrosis Transmembrane Conductance Regulator | ||
|---|---|---|
 | ||
| Darstellung nach 1xmi | ||
|
Vorhandene Strukturdaten: 1NBD, 1XMI, 1XMJ, 2BBO, 2BBS, 2BBT, 2LOB, 2PZE, 2PZF, 2PZG, 3GD7, 3ISW | ||
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 1480 AS; 168 kDa | |
| Sekundär- bis Quartärstruktur | multipass Membranprotein | |
| Isoformen | 3 | |
| Bezeichner | ||
| Gen-Namen | CFTR ; ABC35; ABCC7; CF; CFTR/MRP; MRP7; TNR-CFTR; dJ760C5.1 | |
| Externe IDs | ||
| Transporter-Klassifikation | ||
| TCDB | 3.A.1.202.1 | |
| Bezeichnung | ABC Superfamily | |
| Vorkommen | ||
| Homologie-Familie | ABC-Transporter | |
| Orthologe | ||
| Mensch | Hausmaus | |
| Entrez | 1080 | 12638 |
| Ensembl | ENSG00000001626 | ENSMUSG00000041301 |
| UniProt | P13569 | P26361 |
| Refseq (mRNA) | NM_000492 | NM_021050 |
| Refseq (Protein) | NP_000483 | NP_066388 |
| Genlocus | Chr 7: 117.47 – 117.72 Mb | Chr 6: 18.17 – 18.32 Mb |
| PubMed-Suche | 1080 | 12638 |
Biosynthese
Das CFTR-Gen befindet sich auf Chromosom 7 in der q31.2-Region. Es ist 250 kb lang und besteht aus 27 Exonen. Die transkribierte mRNA hat eine Länge von 6.123 Basen und nach Translation und posttranslationaler Modifikation entsteht das CFTR-Protein mit 1.480 Aminosäuren.[1]
Proteinstruktur
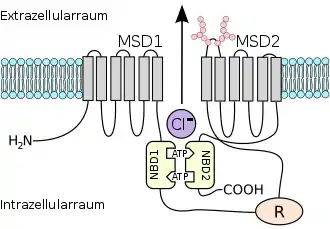
Das Protein ist ein integrales Protein, das zu den cAMP-regulierten Ionenkanälen gehört. Es besitzt zwei Untereinheiten mit je sechs Transmembrandomänen, die den eigentlichen Transportkanal bilden. Zusätzlich besitzt das Protein zwei Nucleotid-Bindedomänen (NBD 1 und 2) und eine cytoplasmische (innerhalb der Zelle befindliche) regulatorische Domäne (R). Diese R-Domäne kann durch die Proteinkinase A phosphoryliert und somit aktiviert werden. CFTR enthält noch weitere Bindedomänen für Protein-Protein-Wechselwirkungen, dadurch reguliert es unter anderem den Transport von Bicarbonat und andere Kanäle. Es ist außerdem ein Mitglied der ATP-bindenden ABC-Transporter.[2]
Funktion und Pathologie
Das CFTR-Protein reguliert den Wasser- und Salztransport in der Plasmamembran von Epithelzellen.
CFTR-Mutationen schränken den Chloridionen-Transport aus der Zelle ein oder bringen ihn zum Erliegen. Folglich entsteht ein Konzentrationsunterschied, da viele Ionen in der Zelle, aber nur wenige im Sekret sind. Aufgrund der in der Zelle vorliegenden osmotischen Kräfte wird dem Sekret Wasser entzogen. Das Sekret wird zähflüssig, kann dadurch schlecht abgebaut werden und verstopft beispielsweise feine Kanäle in der Lunge oder den Vas deferens.
Mutationsklassen
Man unterscheidet sechs Mutationsklassen:
- 1. Klasse: keine Proteinsynthese, da Mutation richtiges Spleißen verhindert
- 2. Klasse: Protein kann nicht im endoplasmatischen Retikulum reifen
- 3. Klasse: Protein reift und erreicht Zielmembran, jedoch fehlende Funktion als Chloridkanal
- 4. Klasse: Protein ist in die Zielmembran eingebaut, hat aber abnormale leitende Eigenschaften
- 5. Klasse: Bildung einiger funktionsfähiger Proteine
- 6. Klasse: Verringerung der Halbwertszeit der Proteine
Klassen 1 bis 3 sind schwere Mutationen, während 4 bis 6 zu den leichten Mutationen zählen – hier ist die Funktion des Kanals nicht vollständig gehemmt.
Ob nun die Mutation zu CF oder CAVD führt, hängt von den Mutationen auf beiden Allelen ab. Der Einfluss anderer Genprodukte spielt eine Rolle bei der Schwere der Krankheit.
ΔF508
Bei dieser Mutation fehlt wegen einer Deletion von drei Nukleotiden an der Stelle 508 die Aminosäure Phenylalanin. Das Protein kann nicht korrekt gefaltet werden, weshalb es von der Proteinqualitätskontrolle im Proteasom abgebaut wird. ΔF508 gehört somit zu den Klasse-2-Mutationen.
R117H
Ist eine etwas mildere aber dennoch häufig auftretende Mutation, sie gehört zu den Klasse-4-Mutationen, die oft bei CBAVD-Patienten gefunden wird. Es ist eine Missense-Mutation, bei der an der 117. Stelle die Aminosäure Arginin durch Histidin ausgetauscht wurde. Diese Mutation führt zu einer geringeren Chloridionen-Leitfähigkeit.
Polymorphismen
Neben den Mutationen sind auch Polymorphismen häufig bei CAVD- und bei CF-Patienten zu finden. Die Polymorphismen gehören zu den Klasse-5-Mutationen. Man hat herausgefunden, dass es Unterschiede gibt in der Thymidinanzahl am Ende der 3'-Spleißstelle vor Exon 9. Man unterscheidet zwischen T5, T7 und T9. Je weniger Thymidin vorhanden ist, desto mehr sinkt die Spleißeffizienz für Exon 9. Dies hat zur Folge, dass die CFTR-Proteine nicht richtig gefaltet sind und somit abgebaut werden. Hier spricht man auch von partieller Penetranz, d. h. Durchschlagskraft. Die partielle Penetranz kann durch einen genetischen Faktor erklärt werden, den (TG)m-Polymorphismus. Denn die Effizienz des Spleißens an Exon 9 hängt u. a. von den TG-Wiederholungen ab. Je mehr TG-Wiederholungen im Intron 8 vorhanden sind, desto ineffizienter ist das Spleißen.
Literatur
- M. Claustres: Molecular pathology of the CFTR locus in male infertility. In: Reproductive Biomedicine Online. Band 10, Nummer 1, Januar 2005, S. 14–41, ISSN 1472-6483. PMID 15705292. (Review).
- H. Cuppens, J. J. Cassiman: CFTR mutations and polymorphisms in male infertility. In: International journal of andrology. Band 27, Nummer 5, Oktober 2004, S. 251–256, ISSN 0105-6263. doi:10.1111/j.1365-2605.2004.00485.x. PMID 15379964. (Review).
- G. Phillipson: Cystic fibrosis and reproduction. In: Reproduction, fertility, and development. Band 10, Nummer 1, 1998, S. 113–119, ISSN 1031-3613. PMID 9727601. (Review).
- W. B. Guggino, B. A. Stanton: New insights into cystic fibrosis: molecular switches that regulate CFTR. In: Nature reviews. Molecular cell biology. Band 7, Nummer 6, Juni 2006, S. 426–436, ISSN 1471-0072. doi:10.1038/nrm1949. PMID 16723978.