Blutzucker-Sensorsystem
Seit langem ist bekannt, dass die Bauchspeicheldrüse (Pankreas) auf die Blutzuckerkonzentration über die Ausschüttung der Peptidhormone Insulin und Glucagon einwirkt (Homöostase). Insulin ist ein Produkt der β-Zellen, Glucagon der α-Zellen des Pankreas.
Neueren Datums sind Kenntnisse über das Blutzuckersensorsystem des Pankreas, d. h. die Grundprinzipien, nach denen Glucosekonzentrationen registriert werden und nachfolgend in eine Ausschüttung von Insulin (Glucosespiegel > 5 mmol/l bzw. 90 mg/dl) bzw. Glucagon (Hungersituation: Glucosespiegel < 4 mmol/l bzw. 70 mg/dl) umgesetzt werden. Das System der β-Zellen gilt heute als weitgehend bekannt, während für jenes der α-Zellen erst einige Komponenten ermittelt werden konnten.
Sensorsystem der pankreatischen β-Zellen
Glucose wird durch einen niederaffinen Glucosetransporter (GLUT-1, Km = 40 mM[1]), proportional zur Blutglucosekonzentration, aufgenommen und über Glucokinase (GK) in den normalen Glycolyse-Weg eingeschleust. Dessen Endprodukt, Pyruvat, wird in den Mitochondrien über Citratzyklus und Atmungskette weiter verstoffwechselt, was letztendlich zur Gewinnung von ATP führt. ATP hat im Folgenden die Funktion eines second messengers: Es leitet das Glucosesignal im Zellinneren weiter, die Zelle reagiert am Ende der Wirkungskette mit der Sekretion von Insulin.
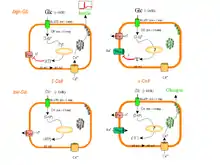
Die Bindeglieder in der Wirkungskette stellen sich wie folgt dar:
- ATP ist ein Inhibitor der ATP-regulierten Kaliumkanäle (K+ATP-Typ). Diese „Kalium-Sickerkanäle“ (Sulfonylharnstoffrezeptor) sind eine der Komponenten, auf die das Membranpotential zurückgeht: das Zellinnere ist um −60 mV negativer eingestellt als die Umgebung
- Inhibition (Schließen) der Kaliumkanäle verringert das Membranpotential. Sobald ein Grenzwert von −40 mV erreicht ist, öffnen sich spannungsgesteuerte Ca++-Kanäle
- Calciumionen strömen von außen ein und bewirken die Wanderung Insulin-haltiger Granula an die Zellmembran, dann die Exozytose von Insulin. Die Insulin-Freisetzung erfolgt in zwei Phasen, d. h., sie beinhaltet zwei Granula-Typen:
- den readily-released pool, der zu einer transienten Insulinspitze führt
- den reserve pool, der 90 % des Insulins gespeichert hat
Bei Glucosemangelsituationen entfällt die Inhibition der Kaliumkanäle in β-Zellen, dafür wird in α-Zellen die komplementäre Situation eingestellt.
Im Falle der β-Zellen sind die bekannten Komponenten in der Abbildung gezeigt, insbesondere die Entstehung und Wirkung des ATP, das hier Second-messenger-Funktionen innehat. Für α-Zellen ist bekannt, dass ATP-regulierte Kaliumkanäle keine entscheidende Rolle spielen. Vielmehr wird über den spannungsabhängigen Natriumkanal ein Aktionspotential ausgelöst, in dessen Folge das Hormon Glucagon sezerniert wird. In beiden Fällen wird Hormonausschüttung aus Granula durch ein Ca2+-Signal ausgelöst.
Sensorsystem der pankreatischen α-Zellen
Auch zum Membranpotential dieses Zelltyps tragen Kalium-Sickerkanäle bei, jedoch handelt es sich hier um Kanäle vom „KA“-Typ. Diese unterliegen nicht der Regulation durch ATP.
Glucosetransport wird hier durch einen hochaffinen Transporter, GLUT-1 (Km = 1 mM) bewirkt, der auf Glucoseschwankungen im unteren Konzentrationsbereich anspricht. Welche Signaltransduktionswege sich in diesem Zelltyp bei Glucoseüberschuss abspielen, ist im Einzelnen nicht bekannt. Jedoch gibt es auch hier Indizien für eine Schlüsselrolle der Glucokinase (GK).
In Glucose-Mangelsituationen wird ein Aktionspotential dadurch ausgelöst, dass das Ruhepotenzial sich vorübergehend erhöht (negativer wird). Dieses Signal führt zur Öffnung der Natriumkanäle und einem unmittelbar folgenden Zusammenbruch des Potenzials auf etwa −40 mV. Zu diesem Zeitpunkt entspricht die Situation jener, der in β-Zellen zur Hormonfreisetzung führt, jedoch ist das Hormon hier der Insulin-Antagonist Glucagon.
Quellen
- Leszek Szablewski: Glucose Homeostasis. In: Glucose Homeostasis and Insulin Resistance. Bentham Science Publishers, 2011. ISBN 978-1-60805-189-2. pp. 46–58.
Einzelnachweise
- Löffler, Georg.: Biochemie und Pathobiochemie. 8., völlig neu bearb. Auflage. Springer, Heidelberg 2007, ISBN 978-3-540-32680-9.