Anomerer Effekt
In der organischen Chemie bezeichnet der anomere Effekt die Tendenz von Atomen in bestimmten Molekülstrukturen eine gewisse räumliche Position bevorzugt einzunehmen, da diese energetisch günstiger ist.
Fachlich genau formuliert nimmt ein Heteroatom, welches an ein Kohlenstoffatom neben einem Heteroatom im Cyclohexanring substituiert ist, bevorzugt die axiale Position gegenüber der (sterisch weniger gehinderten!) äquatorialen Position ein, was sterischen Betrachtungen widerspricht. Anders ausgedrückt ist bei Vorliegen zweier elektronegativer Substituenten an einem Kohlenstoffatom die synclinale Anordnung zweier Bindungen gegenüber der antiperiplanaren bevorzugt. Das Kohlenstoffatom, an das die betreffenden Substituenten gebunden sind, wird als anomerer Kohlenstoff bezeichnet. Im Formelschema ist die Bildung von Molekül 2 mit synclinaler Anordnung der rot markierten Bindungen gegenüber 1 bevorzugt.

Dieser stereoelektronische Effekt tritt auch für jedes der gezeigten Moleküle einzeln auf, da sie in ihrer Sesselkonformation invertieren können. Eine axial stehende Hydroxyfunktion wird damit in eine äquatoriale überführt und umgekehrt; auch hierbei ist diejenige Konformation, in der die OH-Gruppe axial steht, in beiden Fällen bevorzugt.
Der Begriff anomerer Effekt wurde 1958 von Raymond Lemieux und N. J. Chu eingeführt.[1]
Tetrahydropyrane
Der anomere Effekt wurde zuerst in Tetrahydropyranen beobachtet, als man feststellte, dass elektronegative Substituenten (z. B. Alkoxygruppen oder Halogene) in der 2-Position des Tetrahydropyran-Rings die axiale Konformation gegenüber der äquatorialen bevorzugen:[2][3]

Diese axiale Konformation war unerwartet, da sie aufgrund von 1,3-diaxialen Wechselwirkungen verglichen mit der äquatorialen Konformation instabiler sein sollte:[4]

Zum Vergleich beobachtet man in substituiertem Cyclohexan eine Bevorzugung der äquatorialen Konformation des Substituenten, um 1,3-diaxiale Wechselwirkungen zu vermeiden. Hier liegt kein anomerer Effekt vor:[4]

Zucker
Der anomere Effekt besitzt große Bedeutung für die Konformation und Reaktivität von Zuckern.[5] Der anomere Effekt führt in Zuckern zu einer Stabilisierung des α-Anomers,[6] obwohl dieses aufgrund der sterisch ungünstigen axialen Stellung der OH-Gruppe instabil sein sollte.[7] Bei der Synthese von Alkylglucopyranosiden beispielsweise sind die α-Anomere ebenfalls stabiler als die β-Anomere:[8]
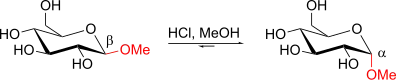
Dies wurde zuerst 1905 von C. L. Jungins entdeckt und 1955 von John T. Edward und 1958 von Raymond Lemieux weiter untersucht, weshalb der anomere Effekt auch Edward-Lemieux-Effekt genannt wird.[9]
Erklärungsansätze
Es gibt mehrere Ansätze zur Erklärung des anomeren Effektes: Dipol-Dipol-Wechselwirkungen, Coulomb-Wechselwirkungen, Mesomerie, negative Hyperkonjugation sowie nichtklassische Wasserstoffbrückenbindungen. Eine Studie aus 2018 zeigt jedoch, dass keine der Erklärungen allein den anomeren Effekt präzise genug begründen kann.[1][10] Vielmehr ist der anomere Effekt ein nicht triviales Zusammenspiel von mehreren korrelierten Wechselwirkungen, die je nach Substrat unterschiedlich ausgeprägt sind. Dabei soll die negative Hyperkonjugation eine geringere Rolle spielen als bisher angenommen und intramolekulare Coulomb-Wechselwirkungen eine bedeutende Rolle spielen. Die negative Hyperkonjugation ist allerdings als Erklärung für den anomeren Effekt in vielen Lehrbüchern der Organischen Chemie anerkannt,[5][6][7][11] ebenso in Lehrbüchern zur Orbitaltheorie.[12][13][14][15]
Dipol-Dipol Wechselwirkungen
John Edward beschrieb 1955 den anomeren Effekt als ein Resultat der Dipol-Dipol-Abstoßung zwischen dem Dipol der C-X Gruppierung (wobei X ein elektronegativer Rest ist) und dem Dipol der C-O-C Gruppierung.[16][17][1] In der äquatorialen Konformation sind die beiden Dipole gleichgerichtet in einer Ebene, wodurch der Dipolcharakter der antiperiplanaren Stellung insgesamt hoch ist. In synclinaler Position heben sich die Dipole partiell auf, was zu einer energetisch niedrigeren und dadurch stabileren, Konformation führt:

Diese Theorie wurde durch die Beobachtung unterstützt, dass das Ausmaß des anomeren Effektes vom Lösungsmittel abhängig ist: In polaren Lösungsmitteln mit großen Dielektrizitätskonstanten ist das polarere Isomer mit dem elektronegativen Rest X in äquatorialer Position stärker stabilisiert als das weniger polarere Isomer, bei dem der Rest axial steht. Polare Lösungsmittel wirken dem anomeren Effekt also entgegen.[1]
Coulomb-Wechselwirkungen
Raymond Lemieux und N. J. Chu schlugen 1971 vor, dass der anomere Effekt ein Ergebnis von Coulomb-Abstoßung ist. Demnach ist die Coulomb-Abstoßung zwischen dem elektronegativen Rest X und dem Sauerstoffatom im Ring in der axialen Konformation geringer als in der äquatorialen Konformation:[1][18]
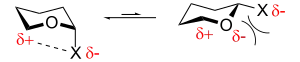
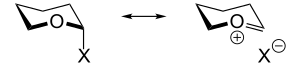
Mesomerie
1969 brachten C. Romers und C. Havinga die Argumentation auf, der anomere Effekt würde (zumindest teilweise) durch Mesomerie mit No-bond Grenzformeln zustande kommen. Diese Mesomerie ist nur in der axialen Konformation möglich:[19]
Negative Hyperkonjugation
Aus Sicht der Molekülorbitaltheorie kann der anomere Effekt als eine Form von negativer Hyperkonjugation erklärt werden.[18] Demnach spendet das nicht bindende 2p-Orbital des Sauerstoffatoms im Ring nur dann Elektronendichte in das σ*-Orbital der C-X Bindung, wenn diese antiperiplanar zum 2p-Orbital des Sauerstoffes steht. Statt des 2p-Orbitals könnte man auch ein sp3-hybridisiertes Orbital am Sauerstoffatom als Donor ansehen. Aus dieser negativen Hyperkonjugation resultiert ein Energiegewinn. Steht der Rest X äquatorial, so findet kein Orbitalüberlapp statt und es resultiert kein Energiegewinn:[1]
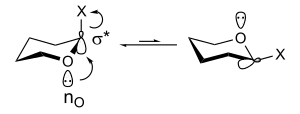
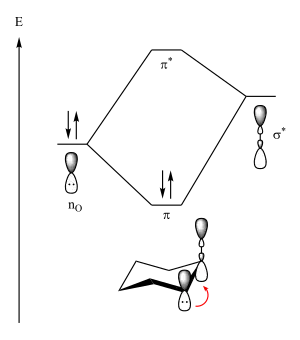
Diese Theorie wird durch die Beobachtung unterstützt, dass Kristallstruktur-Untersuchungen zufolge die C-X Bindung bei Verbindungen mit einem anomeren Effekt verlängert ist, während die C-O Bindung verkürzt ist.[1]
Nichtklassische CH-X Wasserstoffbrückenbindungen
2009 wiesen 0. Takahashi, K. Yamasaki und M. Nishio darauf hin, dass der anomere Effekt durch eine nichtklassische Wasserstoffbrückenbindung zwischen dem axial ständigen elektronegativen Rest X und einem synaxial ständigen Wasserstoffatom begründet werden könnte:[20]

Literatur
- Hans Beyer, Wolfgang Walter: Lehrbuch der organischen Chemie, S. Hirzel Verlag, Stuttgart, 19. Auflage, ISBN 3-7776-0356-2.
- P. Collins, R. Ferrier: Monosacharides - Their Chemistry and their Roles in Natural Products, Wiley West Sussex 1995, ISBN 0-471-95343-1.
Einzelnachweise
- Kenneth B. Wiberg, William F. Bailey, Kyle M. Lambert, Zachary D. Stempel: The Anomeric Effect: It’s Complicated. In: The Journal of Organic Chemistry (Hrsg.): J. Org. Chem. 2018, 83, 5242−5255. 5. April 2018, doi:10.1021/acs.joc.8b00707.
- Theophil Eicher, Siegfried Hauptmann, Andreas Speicher: The Chemistry of Heterocycles: Structures, Reactions, Synthesis and Applications. 3. Auflage. WILEY-VCH Verlag GmbH & Co. KGaA, ISBN 978-3-527-32868-0, S. 317.
- Alan Katritzky, Christopher A. Ramsden, John Joule Viktor Zhdankin: Handbook of Heterocyclic Chemistry. 3. Auflage. Elsevier, ISBN 978-0-08-095843-9, S. 35.
- J. A. Joule, K. Mills: Heterocyclic Chemistry. 5. Auflage. John Wiley & Sons Ltd., ISBN 978-1-4051-3300-5, S. 590.
- Reinhard Brückner: Reaktionsmechanismen. 3. Auflage. Springer Spektrum, ISBN 978-3-662-45684-2, S. 274.
- Jerry March, Michael B. Smith: March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 8th Edition. Hrsg.: Wiley. 8. Auflage. ISBN 978-1-119-37180-9, S. 200.
- H. Beyer, W. Walter, W. Francke, T. Schirmeister, C. Schmuck, P. R. Wich: Organische Chemie. 25. Auflage. Hirzel Verlag, ISBN 3-7776-1673-7, S. 473.
- Pierre Vogel, Kendall N. Houk, Robert H. Grubbs: Organic Chemistry. 1. Auflage. Wiley-VCH, ISBN 978-3-527-34532-8, S. 141.
- Saul Wolfe, Arvi Rauk, Luis M. Tel, I. G. Csizmadia: A theoretical study of the Edward–Lemieux effect (the anomeric effect). The stereochemical requirements of adjacent electron pairs and polar bonds. In: Journal of the Chemical Society B: Physical Organic (Hrsg.): J. Chem. SOC. (B), 1971. doi:10.1039/J29710000136.
- Igor V. Alabugin, Leah Kuhn, Nikolai V. Krivoshchapov, Patricia Mehaffya, Michael G. Medvedev: Anomeric effect, hyperconjugation and electrostatics: lessons from complexity in a classic stereoelectronic phenomenon. Hrsg.: Chemical Society Reviews. 24. August 2021, doi:10.1039/D1CS00564B.
- Jonathan Clayden, Nick Greeves, Stuart Warren: Organische Chemie. 2. Auflage. Springer Spektrum, 2013, ISBN 978-3-642-34715-3, S. 880.
- Albright, Thomas A., Burdett, Jeremy K., Whangbo, Myung-Hwan: Orbital Interactions in Chemistry. Hrsg.: Wiley & Sons Ltd. 2. Auflage. Wiley-VCH, 2013, ISBN 978-0-471-08039-8, S. 233.
- Nguyên Trong Anh: Frontier Orbitals: A Practical Manual. Wiley, 2007, ISBN 978-0-471-97359-1, S. 200.
- Ian Fleming: Molecular Orbitals and Organic Chemical Reactions, Student Edition. John Wiley & Sons, Ltd, 2009, ISBN 978-0-470-74660-8.
- Arvi Rauk: Orbital Interaction Theory of Organic Chemistry. Hrsg.: Wiley-Interscience. 2. Auflage. 2000, ISBN 978-0-471-35833-6, S. 82, 310–311, 305.
- J. T. Edward: Stability of glycosides to acid hydrolysis. A Conformational Analysis. Chem. Ind. (London) 1955, 1102−1104., 1955.
- Ernest L. Eliel, Samuel H. Wilen: Stereochemistry of Organic Compounds. Hrsg.: Wiley-Interscience. 1. Auflage. 1994, ISBN 978-0-471-01670-0, S. 612.
- Claire M. Filloux: The Problem of Origins and Origins of the Problem: Influence of Language on Studies Concerning the Anomeric Effect. In: Angewandte Chemie International Edition (Hrsg.): Angew.Chem.Int. Ed. 2015, 54,8880 –8894. 2015, doi:10.1002/anie.201411185.
- Ernest L. Eliel, Samuel H. Wilen: Stereochemistry of Organic Compounds. Hrsg.: Wiley-Interscience. 1. Auflage. 1994, ISBN 978-0-471-01670-0, S. 612.
- Takahashi, O.; Yamasaki, K.; Kohno, Y.; Ueda, K.; Suezawa, H.; Nishio, M.: The Origin of the Generalized Anomeric Effect: Possibility of CH/n and CH/pi Hydrogen Bonds. In: Carbohydr. Res. 2009, 344, 1225−1229.