Aldolase B
Aldolase B (ALD-B, auch Leber-Aldose oder Fructose-1-phosphat-Aldolase genannt) ist ein Isoenzym der Aldolase, das die Aldolspaltung von Fructose-1,6-bisphosphat sowie von Fructose-1-phosphat katalysieren kann. Die Stoffwechselprodukte der Aldolase B werden in der Glykolyse oder Gluconeogenese weiterverarbeitet.[1]
| Aldolase B | ||
|---|---|---|
| Andere Namen |
| |
| Eigenschaften des menschlichen Proteins | ||
| Masse/Länge Primärstruktur | 364 Aminosäuren, 39.473 Da | |
| Bezeichner | ||
| Gen-Name | ALDOB | |
| Externe IDs | ||
| Enzymklassifikation | ||
| EC, Kategorie | 4.1.2.13 | |
| Orthologe | ||
| Mensch | Hausmaus | |
| Entrez | 229 | 230163 |
| Ensembl | ENSG00000136872 | ENSMUSG00000028307 |
| UniProt | P05062 | Q91Y97 |
| Refseq (mRNA) | NM_000035 | NM_144903 |
| Refseq (Protein) | NP_000026 | NP_659152 |
| Genlocus | Chr 9: 101.42 – 101.45 Mb | Chr 4: 49.54 – 49.55 Mb |
| PubMed-Suche | 229 | 230163 |
Bei Mangel an Aldolase B in der Leber, Niere oder Dünndarm kommt es zur schnellen Anreicherung von Fructose-1-phosphat und kann zur hereditären Fructoseintoleranz führen.[2]
Struktur
Aldolase B ist ein Homotetramer aus 4 β-Unterheiten, das vorwiegend in der Leber exprimiert wird. Die Molmasse des Tetramers beträgt ca. 159 kDa.[3]
Reaktionen
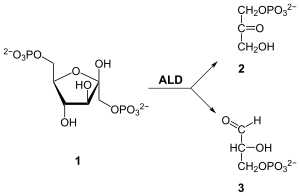

Aldolase B spaltet β-D-Fructose-1-phosphat (2) zu Dihydroxyacetonphosphat (3) und Glycerinaldehyd (4).
Klinische Bedeutung
Bei Mangel an Aldolase B führt eine fructosehaltige Ernährung zur Anreicherung von Fructose-1-phosphat, was mögliche Folgen haben kann:
- Fructose-1-phosphat blockiert die Produktion von Glucose, indem die Gluconeogenese und Glykogenolyse blockiert werden, was zur Hypoglykämie führen kann und ein Symptom der hereditären Fructoseintoleranz ist.[4]
- ATP und Orthophosphat (Pi) werden übermäßig verbraucht, wobei bei Letzterem die Proteinbiosynthese gehemmt wird und zu ultrastrukturellen Läsionen führt, die Fehlfunktionen in der Leber oder Niere verursachen kann.[4]
- Eine mangelhafte Glycosylierung von Proteinen (z. B. Transferrin) durch Hemmung der Mannose-6-phosphat-Isomerase.[5]
Das ALDOB-Gen befindet sich auf Chromosom 9q22.3. Nach der Human Gene Mutation Database am Institute of Medical Genetics in Cardiff sind 25 Mutationen des Gens bekannt, wobei 11 davon zur hereditären Fruktoseintoleranz führen.[6]
Einzelnachweise
- Gerhard Meisenberg, William H. Simmons: Principles of Medical Biochemistry E-Book. Elsevier Health Sciences, 2011, ISBN 978-0-323-08107-8, S. 386 (eingeschränkte Vorschau in der Google-Buchsuche).
- Robert M. Kliegman, Joseph St. Geme: Nelson Textbook of Pediatrics E-Book. Elsevier Health Sciences, 2019, ISBN 978-0-323-56888-3, S. 790 (eingeschränkte Vorschau in der Google-Buchsuche).
- Axel M. Gressner, Torsten Arndt: Lexikon der Medizinischen Laboratoriumsdiagnostik. 3. Auflage. Springer-Verlag, 2019, ISBN 978-3-662-48986-4, S. 62 (eingeschränkte Vorschau in der Google-Buchsuche).
- G. Van den Berghe: Metabolic effects of fructose in the liver. In: Current topics in cellular regulation. Band 13, 1978, S. 97–135, doi:10.1016/b978-0-12-152813-3.50008-2, PMID 208819 (Review).
- J. Jaeken, M. Pirard, M. Adamowicz, E. Pronicka, E. van Schaftingen: Inhibition of phosphomannose isomerase by fructose 1-phosphate: an explanation for defective N-glycosylation in hereditary fructose intolerance. In: Pediatric research. Band 40, Nummer 5, November 1996, S. 764–766, doi:10.1203/00006450-199611000-00017, PMID 8910943.
- HGMD (The Human Gene Mutation Database Cardiff) (Memento vom 2. März 2003 im Internet Archive)