Lymphangioleiomyomatose
Lymphangioleiomyomatose (LAM) ist eine sehr seltene Erkrankung der Lunge, die nahezu ausschließlich bei Frauen auftritt. Sie ist meistens fortschreitend, führt zu chronischem Sauerstoffmangel und ist schließlich lebensbedrohend. In der WHO-Klassifizierung der Lungentumore wird sie auch als mikronoduläre Pneumozytenhyperplasie (MNPH) bezeichnet.
| Klassifikation nach ICD-10 | |
|---|---|
| D48 | Neubildung unsicheren oder unbekannten Verhaltens an sonstigen und nicht näher bezeichneten Lokalisationen |
| D48.1 | Bindegewebe und andere Weichteilgewebe - Lymphangioleiomyomatose |
| ICD-10 online (WHO-Version 2019) | |
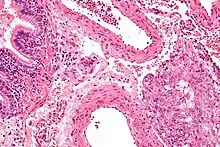
Häufigkeit
Die Krankheitshäufigkeit (Prävalenz) ist nicht bekannt, da keine überregionalen epidemiologischen Daten existieren. Die Erkrankung ist aber insgesamt sehr selten. In Deutschland wird die Zahl der an LAM erkrankten Frauen auf etwa 200 geschätzt. In der größten epidemiologischen Studie zu dieser Erkrankung wurden 243 Patienten in den USA zwischen 1998 und 2001 registriert und befragt. Die Auswertung der Daten ergab unter anderem ein Durchschnittsalter von 41 Jahren zum Zeitpunkt der Diagnosestellung. Der Erkrankungsbeginn lag in etwa 60 % der Fälle vor Eintritt der Menopause, das heißt, bevor bei Frauen die Regelblutungen (Menstruation) aufhören. Die jüngste Patientin war 18, die älteste 76 Jahre alt.[1] Bislang konnte die Krankheit nur bei zwei männlichen Patienten eindeutig nachgewiesen werden.[2][3]
Ursache und Krankheitsentstehung
Die Ursache der Lymphangioleiomyomatose ist nicht vollständig aufgeklärt. Bei der sporadischen Form der LAM liegt eine somatische Mutation des TSC-2-Gens vor. Bei den mit tuberöser Sclerose einhergehenden Fällen ist entweder TSC-1 auf Chromosom 9 oder TSC-2 auf Chromosom 16 mutiert.[4] In den Lungen zerstört ein unkontrolliertes Wachstum der glatten Muskelzellen zunehmend das gesunde Lungengewebe und schränkt damit die Sauerstoffaufnahme des Körpers immer mehr ein. Diese Verwachsungen sind chirurgisch teilweise entfernbar. Dies steigert die Lebensqualität, die Zysten treten jedoch erneut auf.
Der makroskopische Befund[5] ähnelt einem schweren Emphysem mit verbreiterten Alveolarsepten. Dadurch wird das Atmen für Patienten mit LAM immer schwerer und sie sind körperlich wenig belastbar. LAM verursacht bei einem Teil der Betroffenen auch Angiomyolipome der Nieren oder fibrotische Gewebeveränderungen im Bauchraum und vergrößerte Lymphknoten. Bei der Hälfte aller Betroffenen kommt es im Verlauf der Erkrankung zu schweren Symptomen wie Pneumothorax (Kollaps der Lunge) und bei circa 15 % der Frauen Chylothorax (Ansammlung von Lymphflüssigkeit im Pleuraspalt).
Einteilung und klinisches Bild
Die Krankheit gibt es in zwei verschiedenen Formen: einmal als sogenannte sporadische LAM, die nicht vererbt werden kann, und andererseits als LAM, die im Zusammenhang mit der Erkrankung Tuberöse Sklerose auftritt und vererbbar ist. Die ersten Anzeichen der Erkrankung, wie Luftnot bei Belastung oder Husten und Schmerzen im Brustkorb, treten meistens schon im Alter zwischen 25 und 30 Jahren auf. Ein Pleuraerguss kann durch Bildung von Exsudat auftreten.[6] Weil die Lymphangioleiomyomatose so selten ist und meist schleichend beginnt, bleiben die Beschwerden oft ohne korrekte Diagnose oder werden als Asthma oder Lungenemphysem fehldiagnostiziert.
Diagnose
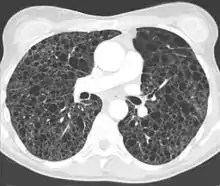
Die eindeutige Diagnose ist mittels Computertomographie oder durch eine Lungenbiopsie möglich. Der CT-radiologische Befund[7] ist typisch und erlaubt in aller Regel die Diagnosestellung.
Therapie
Die bisher gängige Therapie mit Medroxy-Progesteron scheint bei einem Teil der Patienten nur bedingt zu wirken. In der MILES-Studie konnte 2011 gezeigt werden, dass das Medikament Sirolimus (Rapamycin) die Lungenfunktion stabilisiert, die Symptome reduziert und die Lebensqualität verbessert. Nach Absetzen des Medikamentes verschlechtert sich die Lungenfunktion jedoch weiter.[8] Nachdem die Europäische Arzneimittelbehörde (EMA) Sirolimus für die Therapie der sporadischen Lymphangioleiomyomatose (S-LAM) empfohlen hat, besteht aktuell (08/2020) eine Zulassung für die Behandlung von Patienten mit sporadischer Lymphangioleiomyomatose mit mittelschwerer Lungenerkrankung oder abnehmender Lungenfunktion[9]. Viele Betroffene und Ärzte sehen im fortgeschrittenen Krankheitsstadium als einzige Chance die Lungentransplantation, die jedoch nur für jüngere Patienten in gutem Allgemeinzustand infrage kommt. Die Prognose nach einer Lungentransplantation ist gut. Rezidive sind bislang nicht beschrieben.
Einzelnachweise
- J. H. Ryu u. a.: The NHLBI Lymphangioleiomyomatosis Registry: Characteristics of 230 Patients at Enrollment. In: Am J Respir Crit Care Med. 2006 Jan 1;173(1), S. 105–111. PMID 16210669
- M. C. Aubry u. a.: Pulmonary lymphangioleiomyomatosis in a man. In: American journal of respiratory and critical care medicine. Band 162, Nummer 2 Pt 1, August 2000, S. 749–752, ISSN 1073-449X. PMID 10934115.
- M. Schiavina u. a.: Pulmonary Lymphangioleiomyomatosis in a Karyotypically Normal Man without Tuberous Sclerosis Complex. In: American journal of respiratory and critical care medicine. Band 176, Nummer 1, April 2007, S. 96–98, ISSN 1073-449X. PMID 10934115.
- Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. 2012.
- Abbildung aus RadioGraphics (Memento des Originals vom 27. September 2007 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.
- Berthold Jany, Tobias Welte: Pleuraerguss des Erwachsenen – Ursachen, Diagnostik und Therapie. In: Deutsches Ärzteblatt. Band 116, Nr. 21, 2019, S. 377–385, hier: S. 379.
- G. F. Abbott, M. L. Rosado-de-Christenson, A. A. Frazier, T. J. Franks, R. D. Pugatch, J. R. Galvin: From the Archives of the AFIP: Lymphangioleiomyomatosis: Radiologic-Pathologic Correlation. In: Radiographics. 25, 2005, S. 803–828,doi:10.1148/rg.253055006.
- F. X. McCormack u. a.: Efficacy and safety of sirolimus in lymphangioleiomyomatosis. In: The New England Journal of Medicine. Band 364, Nummer 17, April 2011, S. 1595–1606, ISSN 1533-4406. doi:10.1056/NEJMoa1100391. PMID 21410393. PMC 3118601 (freier Volltext).
- Pfizer Europe: Fachinformation Rapamune 0,5 mg/1 mg/2 mg.