Polarografie
Die Polarografie (ein Sonderfall der Voltammetrie) ist ein elektrochemisches Verfahren zur qualitativen und quantitativen Analyse von chemischen Elementen und Verbindungen, vor allem Ionen und Molekülen in einer Lösung. Während bei der Voltammetrie mit stationären Elektroden gearbeitet wird, werden bei der Polarografie Quecksilbertropfelektroden eingesetzt. Sie wurde 1922 von Jaroslav Heyrovský entwickelt und beruht auf der Messung des Elektrolysestroms an einer Quecksilbertropfelektrode. Mit Hilfe der Polarografie ist es möglich, auch unedle Metalle wegen der großen Überspannung von Wasserstoff an Quecksilber bei stark negativen Potenzialen elektrolytisch abzuscheiden und den dabei fließenden Strom zu messen. Dieser stellt das analytische Signal dar.[1]

Aufbau
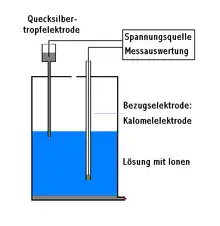
Die Quecksilbertropfelektrode besteht aus einem Quecksilberreservoir und einer Kapillare, aus der Quecksilbertropfen in eine zu untersuchende Lösung fallen. Sie wird als Arbeitselektrode (auch Messelektrode) in der Polarografie verwendet und ist eine ideal polarisierbare Elektrode, d. h., man kann ihr ein elektrisches Potenzial aufprägen, ohne dass es zu einem Ladungsdurchtritt über die Elektrode-Lösung-Phasengrenze kommt, vorausgesetzt in der Lösung befinden sich keine Depolarisatoren. Depolarisatoren sind oxidierbare oder reduzierbare Substanzen. Wenn dies aber doch der Fall ist, kommt es zum Ladungsdurchtritt, die Substanz depolarisiert die Arbeitselektrode, und es fließt ein Strom.
In einer einfachen Zwei-Elektroden-Anordnung übernimmt die Gegenelektrode auch die Funktion der Referenzelektrode. Günstiger ist eine Drei-Elektroden-Anordnung, bei der der Elektrolysestrom über eine Gegenelektrode aus Edelmetall oder Kohlenstoff fließt, während die Referenzelektrode stromlos bleibt. Als Referenzelektrode dient in der Regel eine Elektrode zweiter Art, z. B. eine Kalomelelektrode oder eine Silber-Silberchlorid-Elektrode. Die Vorteile liegen in der längeren Haltbarkeit der Referenzelektrode und geringeren Störungen des angelegten Potenzials durch Überspannungseffekte an der Gegenelektrode.
Messung und Konzentrationsbestimmung
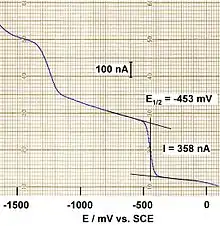
Bei der Messung wird eine zeitlich linear veränderliche Spannung vorgegeben und der entstehende Strom registriert. Wenn ein Stoff in der Lösung eine Durchtrittsreaktion verursacht, kommt es zu einem Stromanstieg, d. h., in der Strom-Spannungs-Kurve tritt eine Stufe auf. Die Lage des Potenzials auf halber Höhe dieser Stufe (Halbstufenpotenzial) ist für jede chemische Spezies charakteristisch, womit eine qualitative Analyse möglich ist. Die Höhe der Stufe (also der Strom) ist durch den Diffusionsgrenzstrom gegeben, der sich dann einstellt, wenn die Diffusion des Analyten aus dem Inneren der Lösung zur Elektrodenoberfläche der geschwindigkeitsbestimmende Reaktionsschritt ist. Dadurch ergibt sich die Möglichkeit zur quantitativen Analyse, da der Diffusiongrenzstrom mit der Konzentration des Analyten über die Ilkovič-Gleichung (eine Zahlenwertgleichung) zusammenhängt:[2]

- : zeitliches Mittel des Stromes, der durch die Diffusion begrenzt wird (mittlerer Diffusionsgrenzstrom) in Mikroampere (µA)
- n: Ladungsanzahl
- D: Diffusionskoeffizient in Zentimeter pro Sekunde (cm2·s−1)
- u: Massefluss des ausfließenden Quecksilbers in Milligramm pro Sekunde (mg/s)
- t : Lebensdauer eines Tropfens in Sekunden (s)
- cL: Konzentration des Analyten (Depolarisators) im Inneren der Lösung in Mol pro Liter (mol/l)

| Temperatur in °C | Dichte von Quecksilber in g/cm³ | Wert des Faktors |
|---|---|---|
| 19,2 | 13,54783 | 606,502 |
| 20,0 | 13,54587 | 606,561 |
| 22,0 | 13,54096 | 606,707 |
| 25,0 | 13,53360 | 606,927 |
| 26,0 | 13,53115 | 607,000 |
| 32,8 | 13,51451 | 607,499 |
Die Gleichung wurde zuerst von Dionýz Ilkovič hergeleitet. Der Faktor K, auch Ilkovič-Konstante[3] genannt, von rund 607 ergibt sich aus der Lösung der Diffusionsgleichung für den wachsenden Tropfen und Mittelung über die Tropfenzeit. Der genaue theoretische Wert ist durch folgenden Ausdruck gegeben[4]:

ρ ist die Dichte des Quecksilbers, F die Faraday-Konstante. An den Beispielwerten in der Tabelle rechts erkennt man, dass der theoretische Zahlenwert 607 zwischen 19,2 °C und 32,8 °C gelten sollte. Das Halbstufenpotenzial und der Diffusionsgrenzstrom sind die charakteristischen Größen für die Art und die Menge des Depolarisators (Analyten) im gewählten Leitelektrolyt. Die Anwendbarkeit der Polarografie wird durch einige Faktoren beschränkt, wie z. B. dem Auftreten eines kapazitiven Stromes, der zu einem Störsignal führt, der die Nachweisgrenze heraufsetzt. Außerdem treten Tropfenzacken und so genannte polarographische Maxima (wenn der Strom aus verschiedenen Gründen über den Grenzstrom ansteigt) auf.
Methoden
Diese Probleme sowie die Anforderung höherer Auflösung und Genauigkeit hat zu verschiedenen verbesserten Polarografiemethoden geführt:
- Rapid-Polarografie: der Tropfen wird mechanisch abgeschlagen
- Tastpolarografie: der Strom wird nur kurz vor Abfallen des Tropfens gemessen
- Derivativpolarografie: statt der Stufenkurve wird die 1. Ableitung dieser Kurve verwendet
- Differenz-Gleichstrompolarografie: Verwendung zweier synchron tropfenden Quecksilbertropfelektroden
- Kathodenstrahlpolarografie: der gesamte in Frage kommende Potenzialbereich wird während eines einzigen Tropfenlebens durchfahren
- Wechselstrompolarografie: der angelegten Gleichspannung wird eine niederfrequente Wechselspannung überlagert
- Pulspolarografie: am Ende des Tropfenlebens wird ein rechteckförmiger Spannungspuls angelegt
- Normalpulspolarografie: Es wird ein Spannungspuls angelegt, der von Tropfen zu Tropfen anwächst. Dazwischen ist die Spannung gleich Null. Jeweils am Ende eines Spannungspulses wird der Strom registriert. Man erhält stufenförmige Signale.
- Differenzpulspolarografie: es wird eine zeitlich linear ansteigende Spannungsrampe gefahren und am Ende eines jeden Tropfenlebens ein konstanter Spannungspuls addiert. Jeweils vor Beginn und vor Ende eines jeden Spannungspulses wird der Strom registriert. Die Differenz aus beiden ergibt den aktuellen Messwert. Man erhält peakförmige Signale.
- Kalousek-Polarografie: einer negativer werdenden Gleichspannung werden positive Rechteckimpulse überlagert bzw. einer konstanten Gleichspannung werden negativer werdende Rechteckimpulse überlagert
- Invers-Voltammetrie: die zu bestimmende Substanz wird an der hängenden Quecksilbertropfenelektrode (HMDE) kathodisch abgeschieden und durch Potenzialdurchlauf in positiver Richtung wieder aufgelöst, wobei dann eine Stromspitze (Peak) auftritt. Sie ist keine Polarografie im eigentlichen Sinne, weil das Quecksilber während der Messung nicht tropft. Stattdessen sind bei der Inversvoltammetrie auch andere Elektrodenmaterialien wie Edelmetalle, Kohlenstoff oder neuerdings auch Bismut in Gebrauch.
Diese Methoden können teilweise weiter unterteilt werden.
Stellenwert

Die Polarografie eignet sich grundsätzlich zur genauen Analyse in einem kleinen Konzentrationsbereich sehr vieler anorganischer und organischer Stoffe. Wegen des großen negativen Potenzialbereiches des Quecksilbers findet dabei überwiegend eine kathodische (reduktive) Umsetzung statt. Die Blütezeit der Polarografie reichte von den 1930er bis in die 1980er Jahre. Sie war die erste breit angewendete instrumentelle Analysemethode. In Form der Atomspektrometrie (Elementanalytik) und der Chromatographie (organische Analytik) erwuchsen in den vergangenen Jahrzehnten bedeutende Alternativverfahren, die sich insbesondere durch eine größere Bandbreite bestimmbarer Analyten auszeichnen.[5]
Vor- und Nachteile
Vorteilhaft sind die hohe erreichbare Genauigkeit (Präzision ca. 1 %), geringe Investitionskosten sowie die Möglichkeit zur Elementspeziesanalyse. In ihrer Abwandlung als Differenzpulspolarografie und inverse Voltammetrie besitzt die Polarografie bei vielen Analyten eine sehr gute Nachweisstärke (vereinzelt die beste aller instrumentellen Methoden, z. B. ppq-Bereich bei Platinmetallen). Der Messbereich kann mehr als 6 Größenordnungen umfassen.[6] Bei der Aufklärung von Redoxreaktionsmechanismen in wässrigen und nichtwässrigen Lösungen kann die Polarografie wertvolle Informationen liefern. Von besonderem Vorteil ist die sich ständig erneuernde und nahezu ideal glatte Elektrodenoberfläche des Quecksilbertropfens.
Nachteilig sind die Störmöglichkeiten durch oberflächenaktive Stoffe, die oftmals geringe Selektivität und der Umgang mit Quecksilber. Letzteres wird zwar vollständig recycelt, beschränkt den Einsatz des Polarografen aber auf das chemische Labor. Obgleich sehr viele Stoffe bestimmt und die meisten Störungen umgangen werden können, setzt die Durchführung der Analysen doch jeweils spezielle Kenntnisse und Erfahrung voraus.
Die Polarografie besitzt noch heute eine große Bedeutung in speziellen Aufgabenbereichen:
Probemedien mit hoher Salzfracht
Galvanische Bäder, Meerwasserproben und Probelösungen aus Schmelzaufschlüssen enthalten hohe Konzentrationen an Alkalimetallsalzen. Diese lassen sich nicht ohne weiteres entfernen. Höhere Salzkonzentrationen stören bei vielen instrumentellen Analyseverfahren wie der Atomspektroskopie. Man kann den störenden Einfluss dieser Probematrix nur durch Verdünnen herabsetzen. Dies verringert jedoch die Nachweisstärke des gesamten Analyseverfahrens. In der Polarografie dienen diese Salze als Grundelektrolyt und stören nicht weiter.
Gelegentliche oder spezielle Untersuchungen
Verglichen mit anderen instrumentellen Methoden ist die Polarografie mit nur geringen Investitionskosten verbunden. Sie bietet Möglichkeiten der Laborautomation, wie zum Beispiel Probenwechsler. Mehrere Hersteller bieten auch heutzutage (2008) moderne computergesteuerte Polarografen an. Daher kann es sich lohnen, bei nur geringem Probeaufkommen statt eines Atomspektrometers einen Polarografen anzuschaffen. Gleiches gilt für ein Chromatografiegerät, falls routinemäßig nur wenige und immer die gleichen organischen Analyten zu bestimmen sind (Qualitätskontrolle).
Siehe auch
Einzelnachweise
- Günter Henze: Polarographie und Voltammetrie. Grundlagen und analytische Praxis. Springer, Berlin u. a. 2001, ISBN 3-540-41394-4, S. 1 f. (eingeschränkte Vorschau in der Google-Buchsuche).
- Ralf Martens-Menzel: Physikalische Chemie in der Analytik. Eine Einführung in die Grundlagen mit Anwendungsbeispielen. Springer, Berlin u. a. 2010, ISBN 978-3-8348-9781-7, S. 169, doi:10.1007/978-3-8348-9781-7 (eingeschränkte Vorschau in der Google-Buchsuche).
- Hans Peter Latscha, Gerald Walter Linti, Helmut Alfons Klein: Analytische Chemie. Chemie-Basiswissen III. 4., vollständig überarbeitete Auflage. Springer, Berlin u. a. 2004, ISBN 3-642-18493-6, S. 354, doi:10.1007/978-3-642-18493-2.
- Klaus J. Vetter: Electrochemical Kinetics. Theoretical and Experimental Aspects. Academic Press, New York u. a. 1967, S. 223.
- Georg Schwedt: Analytische Chemie. Grundlagen, Methoden und Praxis. Thieme, Stuttgart u. a. 1995, ISBN 3-13-100661-7, S. 158.
- Karl Cammann (Hrsg.): Instrumentelle analytische Chemie. Verfahren, Anwendungen und Qualitätssicherung. Spektrum – Akademischer Verlag, Heidelberg u. a. 2001, ISBN 3-8274-0057-0, S. 7–50 f.