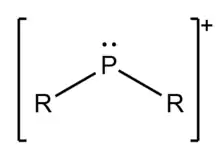
Phosphenium
Als Phosphenium, nicht zu verwechseln mit Phosphonium, werden divalente Phosphorkationen der Form [PR2]+ bezeichnet. Phospheniumionen wurden lange lediglich als Reaktionsintermediate vorgeschlagen, allerdings gibt es auch stabile Derivate.
Synthese
Die ersten cyclischen Phospheniumverbindungen wurden 1972 von Suzanne Fleming und ihren Mitarbeitern synthetisiert.[1] Acyclische Phospheniumverbindungen wurden vier Jahre später, also 1976, erstmals von Flemings Doktorvater Robert Parry beschrieben.[2]
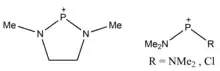
Über die Jahre wurden verschiedene Methoden zur Darstellung von divalenten Phosphorkationen beschrieben. Eine verbreitete Variante ist dabei die Halogenidabstraktion, zum Beispiel mittels Aluminiumtrichlorid von Halogenphosphanen:[3]
- R2PCl + AlCl3 → [R2P+][AlCl4−]
Die Protonolyse von Tris(dimethylamino)phosphan führt ebenfalls zum entsprechenden Phospheniumsalz:[4]
- P(NMe2)3 + 2 HOTf → [P(NMe2)2]OTf + [HNMe2]OTf
Da insbesondere schwach koordinierende Anionen vorteilhaft sind, wird häufig Trifluormethansulfonsäure verwendet.[3]
Weiterhin konnten die zu N-heterocyclischen Carbenen (NHC) isolobalen N-heterocyclische Phospheniumkationen (NHP) synthetisiert werden.[5] So führt die Reaktion von Phosphortriiodid mit einem α-Diimin unter Reduktion des selbigen und Oxidation des Iods zu einem NHP. Gegebenenfalls ist anschließend noch ein Anionentausch notwendig.[5]
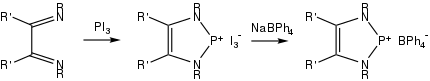
Auch ylidsubstituierte Phospheniumkationen sind bekannt.[6]
Struktur und Bindung
Röntgenstrukturanalytische Daten zeigten, dass [(i-Pr2N)2P]+ nahezu planar mit einem sp2-hybridisierten Phosphorzentrum ist.[7] Die Planarität korreliert mit einer Resonanzstruktur, in der die freien Elektronenpaare des Stickstoffs in Form einer Pi-Bindung Elektronendichte in das vakante 3p-Orbital des Phosphors donieren, dass senkrecht zur N–P–N-Ebene steht. Eine ideal sp2-hybridisierte Verbindung würde einen N–P–N-Winkel von 120° aufweisen, der etwas kleinere Winkel, der in der Molekülstruktur von [(i-Pr2N)2P]+ im Kristall beobachtet werden konnte, lässt sich auf die absoßende Wechselwirkung des freien Elektronenpaars am Phosphor mit den i-Pr2N-Liganden zurückführen. P(NH2)2+ und PH2+ weisen entsprechend Bindungswinkel von 110° beziehungsweise 90° auf.[3][7]
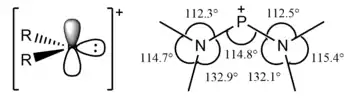
Computerchemische Rechnungen zeigten weiterhin, dass die Analogie zu Carbenen mit stark π-donierenden Substituenten geringer wird. So nehmen NH2-substituierte Phospheniumkationen allylischen Charakter an.[8] Generalized Valence Bond (GVB)-Rechnungen zeigten weiterhin, dass Phospheniumionen einen Singulett-Grundzustand haben. Der Singulett-Triplett-Abstand nimmt weiterhin mit steigender Elektronegativität der Liganden zu.[3][9][10] So beträgt er bei PH2+ 20,38 kcal/mol und bei PF2+ 84,00 kcal/mol. Zusätzlich weist der Triplettzustand einen größeren Bindungswinkel am Phosphor auf. So steigt der berechnete Winkel bei PH2+ von etwa 94° im Singulett- auf 121.5° im Triplettzustand. Die berechneten Bindungslängen beider Strukturen werden hingegen nicht signifikant beeinflusst.[10]
Reaktivität
Phospheniumionen sind isoelektronisch mit Fischercarbenen und entsprechend Lewissauer. Entsprechend führt die Reaktion von [P(NMe2)2]+ und P(NMe2)3 zu Addukten.[2]
- [P(NMe2)2]+ + P(NMe2)3 → [(Me2N)3P-P(NMe2)2]+
Durch ihre elektrophilen Eigenschaften können Phospheniumionen weiterhin in C–H-Bindungen insertieren.[11]
Reaktionen mit Dienen
Phospheniumintermedialte sind Teil der McCormack-Reaktion, einer Methode zur Synthese von Organophosphorheterocyclen. Beispielhaft ist die Reaktion Dichlor(phenyl)phosphan mit Isopren:[12]

Bei isolierten Phospheniumsalzen läuft die Reaktion vollständig ab.[13]
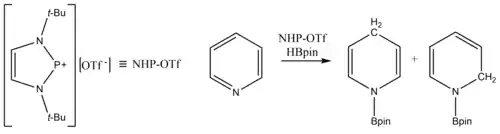
Weiterhin gibt es einige Beispiele für durch Phospheniumionen katalysierte Reaktionen. So berichteten Rei Kinjo und seine Mitarbeiter 2018 die Hydroborierung von Pyridinen durch das NHP-Salz 1,3,2-Diazaphospheniumtriflat. Es wird angenommen, dass das NHP in dieser Reaktion als Hydrid-Transferreagenz dient.[14]
Koordinationschemie

Phospheniumionen dienen als Liganden in der Koordinationschemie.[2] So konnte [(R2N)2PFe(CO)4]+ auf zwei Wegen synthetisiert werden können. Zum einen besteht die Möglichkeit der Fluoridabstraktion von (R2N)2(F)PFe(CO)4 mittels PF5. Weiterhin besteht die Möglichkeit der direkten Substitution eines CO-Liganden von Fe(CO)5 durch das Phospheniumionen.[16] Derartige Fe(CO)4L-Komplexe sind für L= [(Me2N)2P]+, [(Et2N)2P]+, [(Me2N)(Cl)P]+ und [(en)P]+ (en = C2H4(NH2)2) bekannt.[3]

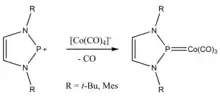
Von N-heterocyclischen Phospheniumion-Übergangsmetallkomplexen wird angenommen, dass sie sich aufgrund der isoelektronischen Eigenschaften wie N-heterocyclische Carbene verhalten. 2004 synthetisierten Martin Nieger und Mitarbeiter zwei Cobalt-NHP-Komplexe. Experimentelle und computerchemische Analysen der untersuchten Komplexe bestätigten die erwartete L→M σ-Hinbindung und M→L π-Rückbindung, wobei das Phospheniumion geringere Donoreigenschaften hatte. Der Grund dafür ist wahrscheinlich der größere s-Orbitalcharakter des freien Elektronenpaars des Phosphors im Vergleich zum analogen Carben.[17] In weiteren Untersuchungen von NHP-Liganden setzten Christine Thomas und Mitarbeitern 2012 Phospheniumionen mit Stickstoffmonoxid um.[18] Stickstoffmonoxid ist dafür bekannt Redoxaktiv zu sein und koordiniert entweder gewinkelt und kann verschiedene L–M-Bindungsmodi eingehen. So stellten sie fest, dass NHPs in Übergangsmetallkomplexen entweder eine planare oder pyramidale Geometrie am Phosphor aufweisen, analog zu der linearen oder gewinkelten Struktur von Nitrosylliganden. Sehr elektronenreiche Metallkomplexe weisen daher ein pyramidales Phosphorzentrum auf, währen elektronenarme Metallkompolexe einen höheren Phospheniumcharakter am Phosphor aufweisen. Die pyramidale Geometrie deutet dabei auf einen signifikanten Lone-Pair-Charakter am Phosphorzentrum hin. Dementsprechend wurden die L→M σ-Hinbindung und die M→L π-Rückbindung durch eine M→L σ-Bindung ersetzt, in der das Metallzentrum formal um zwei Elektronen oxidiert wurde.[18]
Weiterführende Literatur
Cycloadditionsreaktionen
- A. H. Cowley, R. Kemp, J. G. Lasch, N. C. Norman, C. A. Stewart, B. R. Whittlesey, T. C. Wright: Reactivity of phosphenium ions toward 1,3- and 1,4-dienes. In: Inorganic Chemistry. Band 25, 1. März 1986, S. 740–749, doi:10.1021/ic00226a007.
- Carlton K. SooHoo, S. G. Baxter: Phosphenium ions as dienophiles. In: Journal of the American Chemical Society. Band 105, 1. November 1983, S. 7443–7444, doi:10.1021/ja00363a039.
- Michael G. Thomas, Charles W. Schultz, R. W. Parry: Synthesis and characterization of dicoordinate phosphorus cations. Compounds of the type [(R2N)2P]+[Y]- and their congeners. In: Inorganic Chemistry. Band 16, 1. Mai 1977, S. 994–1001, doi:10.1021/ic50171a005.
Addukte
- S. G. Collins, R. L. Collins, A. H. Cowley, S. G. Sena: Ferrocenyl-substituted phosphenium cations and phosphide anions. In: Inorganic Chemistry. Band 22, Nr. 23, 1. November 1983, S. 3475–3479, doi:10.1021/ic00165a022.
- A. H. Cowley, M. Lattman, J. C. Wilburn: NMR study of the reactions of phosphorus(III) halides with halide ion acceptors. Two-coordinate phosphorus cations with bulky ligands. In: Inorganic Chemistry. Band 20, 1. September 1981, S. 2916–2919, doi:10.1021/ic50223a034.
Elektrophile Reaktionen
- A. H. Cowley, R. A. Kemp, C. A. Stewart: Reaction of stannocene and plumbocene with phosphenium ions: oxidative addition of carbon-hydrogen bonds to low-coordination number main group species. In: Journal of the American Chemical Society. Band 104, 1. Juni 1982, S. 3239–3240, doi:10.1021/ja00375a061.
- A. H. Cowley, S. K. Mehrotra: Ring methyl to phosphorus hydrogen shifts in pentamethylcyclopentadienyl-substituted phosphorus cations: parallel between main-group and transition-metal chemistry. In: Journal of the American Chemical Society. Band 105, 1. April 1983, S. 2074–2075, doi:10.1021/ja00345a072.
Koordinationschemie
- V. W. Day, I. Tavanaiepour, S. S. Abdel-Meguid, J. F. Kirner, L. Y. Goh, E. L. Muetterties: Modes of Phosphite Reactions with Transition-Metal Complexes. Crystal Structure of (η5-C5H5)Cr[P(O)(OCH3)2](CO)2[P(OCH3)3]; and [(CH3O)2PMo{P(OCH3)3]5}+(PF6−). In: Inorganic Chemistry. Band 21, 1982, S. 657–663, doi:10.1021/ic00132a038.
- L. D. Hutchins, R. T. Paine, C. G. Campana: Structure and Bonding in a Phosphenium Ion-Metal complex, CH3NCH2CH2N(CH3)PMo(η5-C5H5)(CO)2. An Example of a Molybdenum-Phosphorus Multiple Bond. In: Journal of the American Chemical Society. Band 102, 1980, S. 4521–4523, doi:10.1021/ja00533a039.
- R. W. Light, R. T. Paine: Interaction of the Dicoordinate Phosphorus Cation 1,3-Dimethyl-1,3,2-Diazaphospholidide with Transition Metal Nucleophiles. In: Journal of the American Chemical Society. Band 100, 1978, S. 2230–2231, doi:10.1021/ja00475a043.
- Donn A. Dubois, Eileen N. Duesler, Robert T. Paine: Synthesis and Structure of a Bimetallic Diphosphenium ion Complex Containing a Diazadiphosphetidine Ring. In: Organometallics. Band 2, 1983, S. 1903–1905, doi:10.1021/om50006a044.
- Larry D. Hutchins, Eileen, N. Duesler, Robert T. Paine: Synthesis and Characterization of Metallophosphenium Ion Complexes Derived from Aminohalophosphites. Crystal and Molecular Structure of [cyclo]Mo(η5-C5H5)(CO)2(POCH2CH2NCMe3). In: Organometallics. Band 3, 1984, S. 399–403, doi:10.1021/om00081a013.
Einzelnachweise
- Suzanne Fleming, Mary Kathryn Lupton, Kathleen Jekot: Synthesis of a cyclic fluorodialkylaminophosphine and its coordination with boron acids. Formation of a unique dialkylaminophosphine cation. In: Inorganic Chemistry. Band 11, Nr. 10, Oktober 1972, S. 2534–2540, doi:10.1021/ic50116a050.
- C. W. Schultz, R. W. Parry: Structure of [2((CH3)2N)2PCl].AlCl3, ((CH3)2N)3P.((CH3)2N)2PCl.AlCl3, and related species-diphosphorus cations. In: Inorganic Chemistry. Band 15, Nr. 12, 1. Dezember 1976, S. 3046–3050, doi:10.1021/ic50166a022.
- A. H. Cowley, R. A. Kemp: Synthesis and reaction chemistry of stable two-coordinate phosphorus cations (phosphenium ions). In: Chemical Reviews. Band 85, Nr. 5, 1. Oktober 1985, S. 367–382, doi:10.1021/cr00069a002.
- Otto Dahl: Reactions of aminophosphines with trifluormethanesulfonic acid: phosphenium ion (two-coordinate phosphorus ion) or tricovalent phosphorus products? In: Tetrahedron Letters. Band 23, Nr. 14, Januar 1982, S. 1493–1496, doi:10.1016/S0040-4039(00)87141-5.
- Gregor Reeske, Alan H. Cowley: One-Step Redox Route to N-Heterocyclic Phosphenium Ions. In: Inorganic Chemistry. Band 46, Nr. 4, Februar 2007, S. 1426–1430, doi:10.1021/ic061956z.
- Alfred Schmidpeter, Georg Jochem, Christian Klinger, Christian Robl, Heinrich Nöth: Bis(ylide)-substituted phosphenium and phosphonium halides. In: Journal of Organometallic Chemistry. Band 529, Nr. 1-2, Februar 1997, S. 87–102, doi:10.1016/S0022-328X(96)06443-1.
- Alan H. Cowley, Mike C. Cushner, John S. Szobota: Static and dynamic stereochemistry of dicoordinate phosphorus cations. In: Journal of the American Chemical Society. Band 100, Nr. 24, November 1978, S. 7784–7786, doi:10.1021/ja00492a087.
- Dietrich Gudat: Cation Stabilities, Electrophilicities, and “Carbene Analogue” Character of Low Coordinate Phosphorus Cations. In: European Journal of Inorganic Chemistry. Band 1998, Nr. 8, 1998, S. 1087–1094, doi:10.1002/(SICI)1099-0682(199808)1998:83.0.CO;2-3.
- James F. Harrison, Richard C. Liedtke, Joel F. Liebman: The multiplicity of substituted acyclic carbenes and related molecules. In: Journal of the American Chemical Society. Band 101, Nr. 24, November 1979, S. 7162–7168, doi:10.1021/ja00518a006.
- James F. Harrison: Electronic structure of the phosphenium ions PH2+, HPF+, and PF2+. In: Journal of the American Chemical Society. Band 103, Nr. 25, Dezember 1981, S. 7406–7413, doi:10.1021/ja00415a002.
- Hiroshi Nakazawa, William E. Buhro, Guy Bertrand, J. A. Gladysz: Reactions of phosphorus electrophiles with [(.eta.5-C5Me5)Fe(CO)2]-; spectroscopic evidence for a phosphinidene complex. In: Inorganic Chemistry. Band 23, Nr. 22, Oktober 1984, S. 3431–3433, doi:10.1021/ic00190a001.
- 3-METHYL-1-PHENYLPHOSPHOLENE OXIDE. In: Organic Syntheses. Band 43, 1963, S. 73, doi:10.15227/orgsyn.043.0073.
- A. H. Cowley, R. A. Kemp, J. G. Lasch, N. C. Norman, C. A. Stewart: Reaction of phosphenium ions with 1,3-dienes: a rapid synthesis of phosphorus-containing five-membered rings. In: Journal of the American Chemical Society. Band 105, Nr. 25, November 1983, S. 7444–7445, doi:10.1021/ja00363a040.
- Bin Rao, Che Chang Chong, Rei Kinjo: Metal-Free Regio- and Chemoselective Hydroboration of Pyridines Catalyzed by 1,3,2-Diazaphosphenium Triflate. In: Journal of the American Chemical Society. Band 140, Nr. 2, 17. Januar 2018, S. 652–656, doi:10.1021/jacs.7b09754.
- Alan H. Cowley, Richard A. Kemp, E.A.V. Ebsworth, David W.H. Rankin, Malcolm D. Walkinshaw: Structure/reactivity relationships for cationic (phosphenium)iron tetracarbonyl complexes. In: Journal of Organometallic Chemistry. Band 265, Nr. 2, April 1984, S. c19–c21, doi:10.1016/0022-328X(84)80078-9.
- R. G. Montemayor, Dennis T. Sauer, Suzanne Fleming, Dennis W. Bennett, Michael G. Thomas: Iron carbonyl complexes containing positively charged phosphorus ligands. In: Journal of the American Chemical Society. Band 100, Nr. 7, März 1978, S. 2231–2233, doi:10.1021/ja00475a044.
- Sebastian Burck, Jörg Daniels, Timo Gans-Eichler, Dietrich Gudat, Kalle Nättinen: N-Heterocyclic Phosphenium, Arsenium, and Stibenium Ions as Ligands in Transition Metal Complexes: A Comparative Experimental and Computational Study. In: Zeitschrift für anorganische und allgemeine Chemie. Band 631, Nr. 8, Juni 2005, S. 1403–1412, doi:10.1002/zaac.200400538.
- Baofei Pan, Zhequan Xu, Mark W. Bezpalko, Bruce M. Foxman, Christine M. Thomas: N-Heterocyclic Phosphenium Ligands as Sterically and Electronically-Tunable Isolobal Analogues of Nitrosyls. In: Inorganic Chemistry. Band 51, Nr. 7, 2. April 2012, S. 4170–4179, doi:10.1021/ic202581v.