Osteodystrophia deformans
Die Osteodystrophia deformans, auch als Ostitis deformans, Paget-Syndrom, Paget-Krankheit oder Morbus Paget bezeichnet, ist eine Krankheit des Skeletts mit in einem oder mehreren Arealen erhöhtem und unorganisiertem Knochenumbau, bei der es allmählich zu einer Verdickung der Knochen kommt. Betroffen sind meist Wirbelsäule, Becken, Extremitäten und Schädel. Es handelt sich um eine chronische, langsam fortschreitende Krankheit, an der hauptsächlich ältere Menschen leiden. Sie kann sich auf ein Knochenareal beschränken (monostotisch), oder zwei oder mehr Knochenareale betreffen (polyostotisch). Am Beginn der Krankheitsentwicklung steht eine gesteigerte Aktivität der Osteoklasten, welche Knochensubstanz abbauen. Reaktiv folgen dann ungeordnete Anbauvorgänge, wobei die neue Knochenmasse verformt und brüchig ist. Die Krankheitsursache ist unbekannt. Genetische, virale und Umwelteinflüsse werden diskutiert.
| Klassifikation nach ICD-10 | |
|---|---|
| M88.- | Osteodystrophia deformans |
| M88.0 | Osteodystrophia deformans der Schädelknochen |
| M88.8 | Osteodystrophia deformans sonstiger Knochen |
| M88.9 | Osteodystrophia deformans, nicht näher bezeichnet |
| C41.9 | (Zusatz bei Kombination mit Osteosarkom) |
| ICD-10 online (WHO-Version 2019) | |
Epidemiologie
Die Osteodystrophia deformans tritt häufig in Europa, Amerika, Australien und Neuseeland auf – betroffen sind vorwiegend Menschen europäischer Herkunft. In Asien und Afrika (mit der Ausnahme von Südafrika) ist Osteodystrophia deformans sehr selten. Dieses Verteilungsmuster deute auf genetische Einflüsse und ist konsistent mit der Hypothese einer oder mehrerer Gründer-Mutationen in Nordwesteuropa.
Erstbeschreiber und Namensgeber der Krankheit war 1877 der Pathologe und Chirurg James Paget aus England, wo die Krankheit am häufigsten ist. In Westeuropa sind bis zu 5 % der Männer und 8 % der Frauen im achten Lebensjahrzehnt betroffen, generell Männer häufiger als Frauen. Vor dem 55. Lebensjahr tritt die Erkrankung selten auf, die Prävalenz steigt danach jedoch stetig. Allerdings ist nur ein Bruchteil dieser Patienten klinisch auffällig und behandlungsbedürftig.
Zuletzt wurde ein Rückgang der Inzidenz und der Schwere der Ausprägung des M. Paget beobachtet.
Ätiologie
Der Entstehungsmechanismus der Osteodystrophia deformans ist unbekannt. Da man paramyxovirale RNA, Antigene und Nukleokapside (z. B. Masernviren und Hundestaupeviren)[1][2] in Osteoklasten, Osteoblasten und Osteozyten findet, geht man von einem viralen Einfluss aus. Die Identität der Einschlusskörperchen im Zellkern, die Paramyxovirus-Nukleokapsiden ähneln, konnte jedoch noch nicht zweifelsfrei geklärt werden.
Bei etwa 15 % aller Patienten liegt eine positive Familienanamnese vor, und hierbei zeigt sich ein autosomal-dominanter Erbgang mit unvollständiger Penetranz. Von den Patienten mit positiver Familienanamnese weisen 40 bis 50 % eine Mutation des Gens SQSTM1 (Sequestosome 1, Lokalisation 5q35) auf, das das Protein p62 codiert, welches für die Regulierung der Funktion von Osteoklasten wichtig ist. Auch 5 bis 10 % der Patienten mit einem sporadischen, nicht-familiären M. Paget weisen eine SQSTM1-Mutation auf.
Darüber hinaus gibt es Genmutationen in drei verschiedenen Genen, die fünf seltene klinische Syndrome mit Osteoklasten-Störungen verursachen, die dem Morbus Paget ähnlich sind:
- Insertionsmutationen in Exon 1 des Gens TNFRSF11A (tumor necrosis factor receptor superfamily, member 11a, NFKB activator, Lokalisation 18q22.1), das RANK (auch: CD265) codiert, mit autosomal-dominantem Erbgang, Auftreten im Jugendalter mit ausgedehnten Knochenläsionen, Zahnverlust und Taubheit im Rahmen dreier verschiedener Erkrankungen:
- Familiäre expansile Osteolyse
- Early onset familial Paget’s disease of bone
- Expansive Skeletthyperplasie
- Mutation des Gens TNFRSF11B (tumor necrosis factor receptor superfamily, member 11b, Lokalisation 8q24), das das Protein Osteoprotegerin (früher OPG abgekürzt) codiert, beim Juvenilen Morbus Paget mit Auftreten im Kindesalter, ausgeprägten Knochenläsionen, Knochenbrüchen und Deformitäten, Taubheit sowie vorzeitige Herzkreislauferkrankungen bei autosomal-rezessiver Vererbung
- Mutation des Gens VCP (valosin containing protein, Lokalisation 9p13, Protein p97) im Rahmen eines Syndroms mit Einschlusskörperchen-Myopathie, Morbus Paget und frontotemporaler Demenz, bei autosomal-dominantem Erbgang, Auftreten im dritten bis vierten Lebensjahrzehnt.
Pathogenese
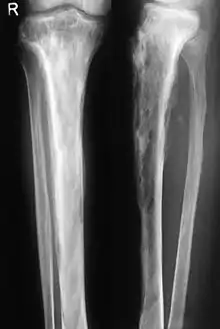
Die Osteodystrophia deformans tritt in der Regel erst jenseits des 55. Lebensjahres auf, verläuft oft symptomlos und wird im Allgemeinen bei einer Untersuchung oder beim Röntgen wegen anderer Beschwerden festgestellt. Sie beginnt als entzündlicher Prozess in einem oder (meistens) mehreren Knochen und ist zu diesem Zeitpunkt schmerzhaft; die Szintigrafie ergibt eine erhöhte Knochenumbaurate. Röntgenbilder des Knochens zeigen unscharfe Aufhellungen.
Im weiteren Verlauf geht die Entzündung zurück und hinterlässt eine dichte, aber unregelmäßige Sklerosierung (kalkreiche Verdichtung) des Knochens, oft auch Deformierungen, druckbedingte Verbiegungen und Auftreibungen der befallenen Skelettelemente, wie eine Wirbelsäulenverkrümmung, einen gewölbter Brustkorb und eine Krümmung der Beine. Bei einer Verdickung der Knochen der Lendenwirbelsäule können Ischiasschmerzen auftreten, die bis ins Bein ausstrahlen (Wurzelkompressionssyndrom).
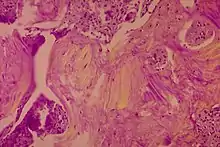
Die Krankheit kann durch lokale Knochenschmerzen auffallen, wobei man durch die Haut die Überwärmung aufgrund der Überaktivität spüren kann. Im fortgeschrittenen Stadium kann es zu einer Verdickung des Schädels mit Zunahme des Kopfumfangs kommen. Sind die Hirnnerven durch das Knochenwachstum geschädigt, kann dies zu Schwerhörigkeit (Versteifung der Gehörknöchelchen, Zuwachsen der Cochlea oder Quetschung des Hörnervs) und Erblindung führen. Feingeweblich ist zu diesem Zeitpunkt ein mosaikähnlich grobmaschiger, aber stabiler lamellärer Reparaturknochen charakteristisch.
Die übermäßige Ausscheidung von Calcium kann zu Nierensteinen führen, die vermehrte Knochendurchblutung begünstigt eine Herzinsuffizienz. Durch die übermäßige Teilungsaktivität der Knochenzellen kommt es in etwa 1 % der Fälle zur Entwicklung eines bösartigen Knochentumors. Wenn derartige Komplikationen vermutet werden, ist eine Computertomografie oder Kernspintomografie sinnvoll.
Diagnose
Bildgebende Verfahren
.jpg.webp)
Wesentlich für die Diagnose ist das Röntgenbild, in dem schon im Frühstadium der Erkrankung die Osteolyse nachgewiesen werden kann. Der erhöhte Knochenumbau kann mittels Knochenszintigraphie nachgewiesen werden.
Laboruntersuchungen
Die Bestimmung der alkalischen Phosphatase (AP) im Serum steht an erster Stelle zur Diagnose und Verlaufsbeobachtung und ist in über 85 % der Fälle bei unbehandeltem Morbus Paget erhöht.[3] Als Ausdruck eines erhöhten Knochenabbaus sind freigesetzte Aminosäuren (vor allem Hydroxyprolin) im Urin nachweisbar. Bedingt durch die vermehrte Aktivität der Osteoklasten ist im Blut ein Anstieg der AP im Serum zu verzeichnen, während die Kalziumwerte daselbst normal bleiben. Marker, die eine erhöhte Knochenresorption (Abbau) des Typ-1-Kollagens beim Knochen anzeigen, wie das C-terminale Telopeptid (CTX), können auch erhöht sein.
Pathologie
Eine Biopsie ist allenfalls in zweifelhaften Frühstadien sinnvoll.
Therapie
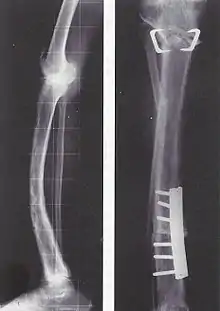
Die Behandlung ist symptomatisch mit schmerzlindernden und entzündungshemmenden Medikamenten wie Nichtsteroidalen Antirheumatika, Entlastung des Knochens, Krankengymnastik und gegebenenfalls operativer Stabilisierung von Knochenbrüchen. Tritt keine Besserung der Symptome ein, können Medikamente den Verlust der Knochenmasse verhindern und Schmerzen lindern; Bisphosphonate und Calcitonin hemmen den Knochenabbau und können bei rechtzeitiger, regelmäßiger Einnahme Deformierungen verhindern. Neuerdings stehen auch Bisphosphonate (Zoledronat, Aclasta®; Pamidronat, Aredia®) zur intravenösen Infusion zur Verfügung. Sie ersetzen die tägliche Tabletteneinnahme. Die einmalige Infusion von Zoledronat hat eine Wirkungsdauer von über einem Jahr. Ergänzend werden Vitamin D und Calcium verschrieben. Bei einer besonders schweren Schädigung der Hüfte kann ein Hüftgelenkersatz erforderlich sein.
Osteodystrophia deformans bei Tieren und im Fossilbericht
Bisher ist diese Erkrankung nur bei wenigen Säugetieren beschrieben worden – so bei Orang-Utans und Lemuren. Die ältesten Menschenknochen, die Anzeichen dieser Erkrankung zeigen, stammen aus der Jungsteinzeit. Im Jahr 2011 wiesen deutsche Forscher die Erkrankung an einem Wirbel des jurassischen Dinosauriers Dysalotosaurus nach. Laut den Forschern handelt es sich dabei um den ältesten indirekten Nachweis einer Vireninfektion.[4]
Literatur
- Wolfgang Piper: Innere Medizin. Springer Medizin Verlag, Heidelberg 2009.
- G. D. Roodman, J. J. Windle: Paget disease of bone. In: J Clin Invest., 2005 Feb;115(2), S. 200–208. Review. PMID 15690073
- J. P. Walsh: Paget’s disease of bone. In: Med J. Aust., 2004 Sep 6, 181(5), S. 262–265. Review. PMID 15347275
- L. Griz, G. Caldas, C. Bandeira, V. Assunção, F. Bandeira: Paget’s disease of bone. In: Arq Bras Endocrinol Metabol. 2006 Aug;50(4), S. 814–822. Review. PMID 17117306
- Leitlinien zur Diagnostik und Behandlung des Morbus Paget des Knochens. (PDF; 216 kB) Dachverband deutschsprachige osteologische Fachgesellschaften
- Stuart H. Ralston: Paget’s Disease of Bone. In: New England Journal of Medicine. 368 (2013), S. 644–650. doi:10.1056/NEJMcp1204713
Weblinks
Einzelnachweise
- M. T. Gordon, D. C. Anderson, P. T. Sharpe: Canine distemper virus localised in bone cells of patients with Paget’s disease In: Bone. 12, 1991, S. 195–201. PMID 1910961.
- J. A. Hoyland, J. A. Dixon, J. L. Berry, M. Davies, P. L. Selby, A. P. Mee: A comparison of in situ hybridisation, reverse transcriptase-polymerase chain reaction (RT-PCR) and in situ-RT-PCR for the detection of canine distemper virus RNA in Paget’s disease. In: J. Virol. Methods. 109, 2003, S. 253–259. PMID 12711070.
- R. Eastell: Biochemical markers of bone turnover in Paget’s disease of bone. In: Bone. 1999; 24(Suppl.5), S. 49–50.
- F. Witzmann, K. M. Claeson, O. Hampe, F. Wieder, A. Hilger, I. Manke, M. Niederhagen, B. M. Rothschild, P. Asbach: Paget disease of bone in a Jurassic dinosaur. In: Current Biology. 2011; 21(17), S. R647–R648 (13 September 2011). (PDF) (Memento des Originals vom 6. Juni 2013 im Internet Archive) Info: Der Archivlink wurde automatisch eingesetzt und noch nicht geprüft. Bitte prüfe Original- und Archivlink gemäß Anleitung und entferne dann diesen Hinweis.