Neurobiologische Schizophreniekonzepte
Unter dem Stichwort neurobiologische Schizophreniekonzepte werden Befunde und Theorien zusammengefasst, die sich mit den vornehmlich von Naturwissenschaftlern erstellten Modellen der Schizophrenie als Krankheit beschäftigen. Die moderne Medizin erforscht Ätiologie und Pathogenese der Schizophrenie mit großem Aufwand und führt Studien in vielen Bereichen durch.[1] Die hier vorgestellten betreffen in erster Linie Genetik, Neurochemie, Neuropharmakologie, morphologische Befunde und sonstige organische Faktoren.
Genetik
Historische Aspekte
Die Tradition der modernen genetischen Forschung im Bereich der Schizophrenie geht auf den umstrittenen deutschen Genetiker Ernst Rüdin[2][3] zurück. Rüdin war lange Jahre Leiter der Deutschen Forschungsanstalt für Psychiatrie (kurz DFA), der Vorläuferorganisation des Max-Planck-Institutes für Psychiatrie in München. Die DFA wurde 1917 auf Initiative von Emil Kraepelin gegründet und 1924 der Kaiser-Wilhelm-Gesellschaft angegliedert. Rüdin war ab 1918 Leiter der Genealogisch-Demographischen Abteilung der DFA und ab 1931 geschäftsführender Direktor der gesamten DFA. Er ist durch seine einflussreiche Mitarbeit an dem so genannten Gesetz zur Verhütung erbkranken Nachwuchses von ärztlicher Seite hauptverantwortlich für die Zwangssterilisation von mehreren hunderttausend Menschen in der Zeit des Nationalsozialismus. Die argumentative Grundlage für diese Gesetzgebung und ihre kriminelle Praxis waren unter anderem die von Rüdin und Anderen angestellten empirischen Untersuchungen über die Vererblichkeit seelischer Erkrankungen. Rüdin gilt hier als ein zu dieser Zeit international anerkannter Pionier.[4]
Populationsgenetische Studien
Die grundlegendsten Aussagen zur Populationsgenetik der Schizophrenie betreffen die familiäre Häufung, Zwillingsstudien und Adoptionsstudien der Erkrankung. Zwar treten 80 % der Schizophrenien sporadisch auf, das Erkrankungsrisiko ist jedoch bei Verwandten ersten Grades (Eltern, Kinder, Geschwister) schizophren Erkrankter deutlich erhöht.
| Zeitraum | Anzahl der Studien |
Verwandte | Inzidenz |
|---|---|---|---|
|
|
|
|
Bei einem Lebenszeitrisiko von ca. 1 % für die Durchschnittsbevölkerung beträgt das Risiko eines Geschwisters eines Schizophrenen etwa 10 %, das eines zweieiigen Zwillingsgeschwisters ca. 14 % und das eines eineiigen Zwillingsgeschwisters etwa 46 %. Die Konkordanzraten für eineiige Zwillinge sind hoch, aber noch weit unterhalb von hundert Prozent, was für eine genetische Komponente der Erkrankung spricht, aber auch dafür, dass Umweltfaktoren zur Verursachung der Schizophrenie beitragen.
| Studie | Übereinstimmende MZ-Paare |
Übereinstimmende DZ-Paare |
|---|---|---|
|
|
|
Die Adoptionsstudien zur Schizophrenie haben schließlich gezeigt, dass das Erkrankungsrisiko nicht vom Erziehungsstil der leiblichen oder Pflegeeltern abhängt.
| Probanden | Schizophreniefälle unter biologischen Verwandten |
Schizophreniefälle in der Adoptivfamilie |
|---|---|---|
|
|
|
Diese Daten zeigen von Studie zu Studie teilweise große Unterschiede, die vermutlich im Wesentlichen auf das Studiendesign zurückgehen. Alle Arbeiten zur Frage der familiären Häufung der Schizophrenie zeigen aber einen Trend zu einem deutlich erhöhten Risiko in Abhängigkeit zum Verwandtschaftsgrad. Der Vererbungsmodus der Schizophrenie ist unklar. Ob ein Unterschied zwischen der familiären gegenüber der sporadischen Form der Schizophrenie besteht, ist nicht gesichert.[9]
Humangenetik der Schizophrenie
Zur Frage der molekularen Genetik der Schizophrenie wurden zahlreiche Studien durchgeführt. Die genetischen Studien beruhen im Wesentlichen auf zwei Ansätzen: Koppelungsstudien und Assoziationsstudien. Das Prinzip der Kopplung beruht darauf, dass ein „Krankheitsgen“ mit einem „Markergen“ gekoppelt ist. Das heißt einfach, dass die entsprechenden Gene auf einem Chromosom eng benachbart sind. Kopplungsanalysen lassen sich sinnvoll bei Erkrankungen mit mendelschem Erbgang einsetzen. Bei Assoziationsstudien sucht man nach beliebigen Sequenzvarianten, die mit einem Merkmal gemeinsam vererbt werden. Diese Art von genetischen Studien wird bei Untersuchungen bevorzugt, bei denen man prüfen will, ob ein vermutetes Kandidatengen mit einer Erkrankung im Zusammenhang steht. Ein genetischer Zusammenhang mit einer Erkrankung kann aber auch bei einem nichtcodierenden Genabschnitt bestehen. Assoziationsstudien haben eine geringere Aussagekraft als Koppelungsstudien. Zur Übersicht:[10][11][12]
Eine Übersicht der jüngsten Arbeiten zur Genetik der Schizophrenie ergibt, dass 6 Gene bzw. Genregionen als aussichtsreiche Kandidaten für ein „Schizophrenie-Gen“ gehandelt werden:
- Das Dysbindin-Gen (DTNBP1) liegt auf dem Chromosom 6p22. 3. Es findet sich vor allem im Bereich des Kleinhirns und des Hippocampus in postsynaptischen Strukturen. Es ändert möglicherweise die präsynaptische Funktion von Glutamat. Bei Schizophrenen gibt es Hinweise für eine reduzierte Expression des Dysbindin-Gens.
- Das Gen für Neuregulin 1 (NRG-1) liegt auf Chromosom 8p21. Das NRG-1 Gen ist sehr groß (über 1 Mb) und codiert für mehr als 15 Proteine. Es enthält alleine 6 Regulationsgene. Möglicherweise ist bei Schizophrenen eine Mutation im Regulationsgen IV des NRG-1 für eine veränderte Expression dieses NRG-1 Subtyps verantwortlich. Einige jüngere Studien legen nahe, dass Veränderungen von NRG-1 und seinem Rezeptor ErbB4 das Risiko, an einer Schizophrenie zu erkranken, erhöhen.[13]
- Das Gen für DISC1 wurde bei einer Familie mit Schizophrenie identifiziert, bei der man eine balancierte Translokation t (1, 11)(q42; q14. 3) gefunden hat. In diesem Bereich werden durch die Translokation zwei Gene zerstört: DISC1 und DISC2. DISC2 enthält keine kodierenden Abschnitte. Es regelt aber möglicherweise die Expression von DISC1 durch Bildung einer spezifischen Antisense-RNA. DISC1 ist möglicherweise verantwortlich für Prozesse der neuronalen Migration, da es an Anteile des neuronalen Zytoskeletts bindet.
- Das Gen für DAOA (D-amino acid oxydase activator, früher G72) liegt auf Chromosom 13q22. 34. DAOA wird nur bei Primaten im Bereich des Nucleus caudatus und der Amygdala exprimiert. Es aktiviert, wie der Name sagt, DAO (D-amino acid oxydase), die D-Serin oxidiert, welches wiederum ein Aktivator des NMDA-Glutamat-Rezeptors ist. Bei verschiedenen Studien wurde gefunden, dass einige DAOA-Polymorphismen mit einem erhöhten Risiko für Schizophrenie einhergehen. DAOA gilt als ein „schwächerer“ Kandidat, also NRG-1 und DISC 1.
- Das Gen für COMT (Catechol-O-Methyltransferase) liegt auf dem Chromosom 22q11. COMT hat eine Schlüsselrolle im Metabolismus der Katecholamine. Es baut im synaptischen Spalt Dopamin zu Homovanillinsäure und Methoxythyramin ab. Es gibt zwei Formen von COMT: eine lösliche Form (S-COMT) und eine membrangebundene Form (MB-COMT). Findet sich bei S-COMT im Codon 108 statt eines Methionins ein Valin und bei MB-COMT in Codon 158 statt eines Methionins ein Valin, dann geht dies mit einer erhöhten thermischen Stabilität des Proteins einher. Man vermutet, dass Träger solcher Allele eine stabilere und also auch aktivere Form des COMT besitzen und daher bei ihnen Dopamin besser abgebaut wird. Verschiedene Studien haben gezeigt, dass das Vorliegen der Valin-Variante mit einem erhöhten Risiko für Schizophrenie einhergeht. Dieser Befund würde zur Hypofrontalitätsthese der Schizophrenie passen. Die Ergebnisse der Assoziationsstudien zu COMT/Schizophrenie sind allerdings sehr widersprüchlich.
- Das Gen für RSG4 liegt auf Chromosom 1q22. Es ist ein negativer Regulator von G-Protein-gekoppelten Rezeptoren. RGS4 wird durch dopaminerge Aktivität reguliert und regelt selbst wiederum die Aktivität von serotoninergen und glutamergen Neuronen. Es interagiert mit ErbB3, das ein Rezeptor von NRG1 ist.
Eine Zusammenfassung der Daten ist in der folgenden Tabelle gegeben:
| Autor | Dysbindin | Neuregulin | DAOA | DISC 1 | COMT | RGS 4 |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
Von allen genannten Kandidatengenen gelten Dysbindin und NRG1 als aussichtsreichste Gene. Allerdings werden noch zahlreiche weitere Kandidaten in den diversen Übersichtsarbeiten genannt. Zu ihnen zählen der metabotrope Glutamatrezeptor GRM-3 auf Chromosom 7q, die Glutamat-Decarboxylase 1 auf Chromosom 2q und ein virales Oncogen AKT1, das bei Mäusen Thymome induziert. Eine Auswahl weiterer Befunde kann angeschlossen werden:
- Als guter Kandidat galt die Region 1. 4 im Chromosom 1, die aufgrund einer partiellen Trisomie 5 bei einer Familie mit zwei schizophren Erkrankten festgestellt wurde.[20]
- Bei einer Reihe von Familien mit schizophren Erkrankten fand sich eine partielle Translokation des Chromosoms 11 in der Nähe der Genregion, bei der sich Gene für den D2-Rezeptor, Tyrosinkinasen und NCAM (neuronales Zelladhäsionsmolekül) fanden.
- Es gibt einen fraglichen Zusammenhang eines Polymorphismus des 5-HT2A-Rezeptors mit einem bevorzugten Ansprechen auf Clozapin.
- Marker auf dem Chromosom 18p betreffen möglicherweise schizophrene und affektive Psychosen.[21][22]
Wegen der Vielzahl von Markern, die für die Schizophrenie auf praktisch allen Chromosomen mit Ausnahme der Nummer 23 gefunden wurden, bezweifeln inzwischen immer mehr Wissenschaftler, ob die bisherigen Untersuchungsstrategien für die Genetik der Schizophrenie zu einem Erfolg führen werden. Die amerikanische Genetikerin Lynn DeLisi hat deshalb vorgeschlagen, nicht mehr nach krankheitsassoziierten Mutationen zu suchen. Sie schlug vor, den Methylierungsstatus des X-Chromosoms bei Patienten mit einem Klinefelter-Syndrom und Schizophrenie zu untersuchen. Die Vorstellung ist dabei, dass eine fehlerhafte Inaktivierung bestimmter Gene des X-Chromosoms zum Risiko für die Entstehung der Schizophrenie beiträgt. Zu diesem Zweck haben DeLisi und andere den Methylierungsstatus von X-chromosomalen Genen untersucht, die nur beim Menschen vorkommen.[23]
Neurochemie und Neuropharmakologie
Es liegt nahe, für die psychopathologischen Phänomene ähnliche neuronale Ursachen anzunehmen wie für die normalen psychischen Funktionen. Allerdings ist aufgrund der Vielfalt der Symptome der Schizophrenie nicht davon auszugehen, dass es ein spezifisches neurochemisches Störungsmuster gibt. Seit der Entdeckung der Neuroleptika konzentriert sich ein großer Bereich der wissenschaftlichen Ursachenforschung zur Schizophrenie auf die Frage, welche Bedeutung das dopaminerge System im Gehirn des Menschen für die Entstehung der Schizophrenie hat. In den letzten Jahren werden zunehmend auch andere Transmittersysteme untersucht. Dies hat unter anderem seine Ursache in der Entdeckung der so genannten atypischen Neuroleptika.
Dopamin
Das Wirkprinzip der klassischen Neuroleptika ist die Blockade der Dopaminrezeptoren im Gehirn. Dadurch kommt es zu einer veränderten Aktivität der durch das dopaminerge System versorgten Nervenzellverbände.
Anatomie der dopaminergen Systeme
Es gibt im menschlichen Gehirn vier dopaminerge Systeme. Das nigrostriatale System ist eine Verbindung dopaminerger Neuronen aus dem Hirnstamm zu den Basalganglien. Störungen des nigrostriatalen Systems führen bei der Parkinsonerkrankung zu Bewegungsstörungen. Bei der Einnahme von Neuroleptika kommt es zu ähnlichen Symptomen. Das tuberoinfundibuläre System besteht aus dopaminergen Neuronen, die die Prolaktinsekretion regeln. Die Einnahme von Neuroleptika führt nicht selten zu einer Erhöhung des Prolaktins im Serum und entsprechender Nebenwirkungen. Das mesolimbische System ist für die Regulation von Affekten verantwortlich. Die mesofrontocorticalen und mesohippocampalen Systeme werden für Prozesse im Bereich von Kognition und Gedächtnis verantwortlich gemacht.
Dopamin und Dopaminrezeptoren
Es gibt zwei verschiedene Dopaminrezeptorfamilien, die mit den Abkürzungen D1 und D2 bezeichnet werden. Die D1-Familie enthält die zwei Subtypen D1 und D5. Die D2-Familie enthält die drei Subtypen D2, D3 und D4. Für die antipsychotische Wirkung der Neuroleptika sind hauptsächlich die D2-Rezeptoren verantwortlich. Messungen der Dopaminkonzentrationen bei schizophren erkrankten Menschen ergaben sehr widersprüchliche Ergebnisse. Vermutlich trägt der Dopaminüberschuss, der eine Schizophrenie verursacht, nicht wesentlich zu den messbaren Konzentrationen bei. Der Dopaminmetabolit Homovanillinsäure kann im Liquor gemessen werden. Seine Konzentration korreliert mit der Einnahme von Neuroleptika. Bei schizophrenen Patienten wurde in post-mortem-Studien eine vermehrte Anzahl von D2-Rezeptoren im Gehirn gefunden, was nach heute übereinstimmender Meinung durch die Einnahme von Neuroleptika verursacht ist. Zahlreiche Studien befassten sich mit dem Nachweis von an D2-Rezeptoren gebundenen radioaktiv markierten Neuroleptika bei Patienten und Probanden. Die Ergebnisse dieser Studien lassen folgende Schlussfolgerungen zu: bei den üblichen Dosierungen typischer Neuroleptika werden 70 – 80 % der Rezeptoren blockiert. Es gibt dabei keine Unterschiede zwischen Respondern und Non-Respondern. Klassische Neuroleptika blockieren dabei auch D1-Rezeptoren, atypische unter anderem auch Serotonin-Rezeptoren.
Dopaminhypothese der Schizophrenien
Carlson und Snyder postulierten vor über vierzig Jahren die Hypothese, dass psychotische Symptome durch einen Überschuss an Dopamin verursacht werden.[24][25][26] Eine Blockade der Dopaminrezeptoren wird dann wie im Falle der Neuroleptika psychotische Symptome mildern. Andererseits kann die Einnahme von Amphetamin euphorisierend wirken und bei längerer Einnahme Psychosen auslösen.[27][28] Amphetamin bewirkt eine Freisetzung von Dopamin und hemmt die Inaktivierung desselben. Amphetaminpsychosen sprechen sehr schnell auf die Gabe von Neuroleptika an. Diese Beobachtungen stützen die Dopaminhypothese. Als Probleme der Dopaminhypothese wurden diskutiert:
- Schizophrene Minussymptome werden durch eine Dopaminblockade verstärkt. Aus diesem Grund postulierte Tim Crow die Existenz zweier verschiedener Schizophrenieformen (Typ I und Typ II).[29]
- Die klinische Wirkung der Neuroleptika setzt nicht so schnell ein wie die pharmakologische Wirkung.
Nach Verabreichung einer ausreichenden Dosis eines Neuroleptikums sind spätestens nach zwei Stunden alle Dopaminrezeptoren besetzt. Die antipsychotische Wirkung setzt aber häufig erst ein, wenn ein Neuroleptikum über Tage oder gar Wochen eingenommen wird. Deshalb vermutet man als antipsychotischen Wirkmechanismus nicht die Rezeptorblockade selbst, sondern den verzögert einsetzenden Depolarisationsblock.[30]
Glutamat
Seit über 15 Jahren wird auch eine Glutamat-Hypothese der Schizophrenie diskutiert. Ein starkes Argument für diese Hypothese ist die Existenz eines analogen Phänomens zur Amphetaminpsychose, der Glutamatpsychose durch Phencyclidin (PCP). Die psychoseauslösende Wirkung des Phencyclidins ist seit langem bekannt. Vor allem das L-Isomer des PCP-Derivates Ketamin, das in der Tiermedizin und früher auch in der Kinderheilkunde für Narkosen eingesetzt wurde, kann akute Psychosen auslösen. PCP kann bei gesunden Probanden nicht nur Positiv-, sondern auch Negativ-Symptome auslösen. Die PCP-Psychose gilt daher als ideales Modell für die Schizophrenie.[31]
Zur Neuroanatomie der glutamergen Neurone ist zu bemerken, dass es sich beim Glutamat um den wichtigsten Neurotransmitter der corticalen Neurone handelt. Es sind bislang acht glutamerge Rezeptoren identifiziert worden. Sie teilen sich in zwei Gruppen: drei ionotrope und fünf metabotrope Rezeptoren. Von allen Glutamat-Rezeptoren ist der NMDA-Rezeptor der psychiatrisch interessanteste. Er wird seit über 20 Jahren intensiv untersucht.[32] Zur Übersicht vergleiche:[33]
Serotonin (5-HT)
.svg.png.webp)
Das serotonerge System hat elementaren Einfluss auf Wahrnehmung und Empfinden. So ist die Halluzinogenesis „klassischer“ psychedelischer Halluzinogene, wie beispielsweise Meskalin, auf serotonerge Mechanismen zurückzuführen. Zentrale Bedeutung kommt dabei dem 5-HT2A-Rezeptor zu.[34] So zeigen Arzneistoffe, die bevorzugt den 5-HT2A-Rezeptor hemmen (MDL 100907), eine antipsychotische Wirkung. Weitere Serotonin-Rezeptoren stehen in der Diskussion, an der bewusstseinsverändernden Gesamtwirkung einiger Halluzinogene mit beteiligt zu sein, darunter 5-HT1A,[35] 5-HT5A, 5-HT7. Dennoch fällt es schwer, aus diesen Erkenntnissen ein schlüssiges Erklärungsmodell der Schizophrenie herzuleiten. Denn schizophrene Patienten erleben die durch serotonerge Halluzinogene erzeugten Psychosen anders als die ihnen vertrauten Krankheits-Symptome. (H. Leuner) Dies schränkt den Wert der „LSD“-Psychose als Modell für die Schizophrenieforschung ein. Osmond und Symythies stellten 1952 die Transmethylierungshypothese der Schizophrenie auf.[36] Diese besagt, dass körpereigene Substanzen in psychose-auslösende Stoffe, ähnlich dem LSD, umgebaut würden. Diese Hypothese gilt als nicht belegt.
Sonstige neurochemische Befunde
Vorläufige Studien ergaben bei Post-mortem-Untersuchungen Hinweise auf eine Erhöhung des Anteils von Dopamin-Rezeptor-Dimeren bei Schizophrenie.[37]
Morphologische Befunde
Anfänge
Neuropathologische Untersuchungen bei Menschen, die an einer Schizophrenie erkrankt waren, datieren aus der Zeit des Beginns der Hirnforschung. In den 1890er Jahren gründete Emil Kraepelin ein neuroanatomisches Labor an der Universität München. Seine Schüler Alois Alzheimer, Robert Gaupp und Franz Nissl begannen dort mit ihren Studien zur Neuropathologie der Schizophrenie.
Alzheimer publizierte 1897 eine der ersten Arbeiten zu diesem Thema.[38] Bis Mitte des 20. Jh. wurden zahlreiche Studien zu diesem Thema veröffentlicht, die aber keine einheitlichen Ergebnisse vorweisen konnten. Daher wurde über einen Zeitraum von mehr als 30 Jahren kaum in diesem Bereich publiziert.[39]
Pneumenzephalographie
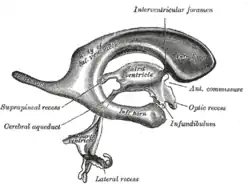
Der Beginn der modernen Forschungen zu den morphologischen Veränderungen bei Schizophrenien bilden die bereits in den 1950er-Jahren von Gerd Huber durchgeführten pneumenzephalographischen Untersuchungen. Dabei entdeckte Huber die Ventrikelasymmetrie bei Schizophrenen.[40] Bei der Pneumenzephalographie wird der Liquor großenteils gegen Luft ausgetauscht und es werden konventionelle Schädel-Röntgenaufnahmen gemacht. Die Luft in den inneren und äußeren Hohlräumen des Gehirns bewirkt einen Kontrasteffekt, der die Umrisse des Gehirns und je nach Lagerung des Patienten Teile des Ventrikelsystems plastisch dargestellt erscheinen lässt.[41] Dieses Verfahren wurde in der Ära vor der Computertomographie unter anderem zur Diagnose von Hirntumoren eingesetzt und zur Planung stereotaktischer Operationen.
CCT und MRT
Seit der Erfindung der Computertomographie und der ersten CT-Studie bei schizophrenen Patienten durch Johnstone[42] wurden über 200 computertomographische und kernspintomographische-Studien bei diesen Patienten durchgeführt. Diese Untersuchungen haben bewiesen, dass Menschen mit einer Schizophrenie links betont erweiterte Seitenventrikel haben. Die Untersuchungsbefunde deuten darauf hin, dass es unter den an Schizophrenie erkrankten Menschen mit einer Ventrikelasymmetrie keine weiteren Untergruppen mit einem speziellen Typ von Ventrikelerweiterung gibt. Es gibt zudem keine eindeutige Korrelation der Ventrikelweite zu einem spezifischen Symptom. Zudem ist dieses Phänomen nicht schizophrenie-spezifisch, es findet sich auch bei Patienten mit affektiven Störungen. Die Ventrikelerweiterung scheint genetisch determiniert zu sein. Zumindest findet sich bei den Patienten keine Korrelation zu einem anderen untersuchten Merkmal wie Alter, Geschlecht, Behandlung, soziale Gruppe etc. Allerdings finden sich die Ventrikelerweiterungen auch bei den nahen Verwandten der Patienten. Bislang gibt es noch keine Ergebnisse von prospektiven Studien zur Frage des Erkrankungsrisikos bei Ventrikelerweiterung. Die Ursache des Phänomens liegt vermutlich in einer Verringerung der Anzahl der Zellen, die den Ventrikelräumen benachbart sind. Teilweise geht die Ventrikelerweiterung auf das Konto von Volumenminderungen im Bereich des Hippocampus. Zur Übersicht:[43]
Volumetrische Studien bei Schizophrenie
- Gesamtvolumen
Es gibt bis zum Jahr 2001 23 Studien, in denen Veränderungen des Gesamtvolumens des Gehirns von schizophrenen Patienten untersucht worden ist.[44] Dabei zeigte sich, dass es in der Mehrzahl der Fälle keine signifikanten Veränderungen gibt. Ein Teil der Patienten zeigte eine Abnahme des Gehirnvolumens.
- Volumenveränderungen in grauer und weißer Substanz.
Untersuchungen zu Veränderungen des Volumens von grauer und weißer Substanz haben deutliche Veränderungen gezeigt. In der überwiegenden Mehrzahl der untersuchten Fälle in insgesamt 20 Studien bis zum Jahr 2000 zeigte sich eine Volumenreduktion der grauen Substanz, wobei die gemessenen Volumen der weißen Substanz unverändert waren.[45] Einzelne Studien haben einen Effekt der Volumenminderung auf die Wirkung von Neuroleptika gezeigt.[46][47]
- Ventrikelvolumen
Eine Erweiterung der Seitenventrikel bei Schizophrenie ist der am häufigsten replizierte Befund. Dies konnte in einer Zusammenstellung von 28 Studien aus den Jahren 1994 bis 2000 in über 95 % der Fälle nachgewiesen werden.[48] Die Ursachen der Ventrikelasymmetrie ist nicht genau bekannt. In einzelnen Studien konnte ein genereller Hirngewebsverlust nachgewiesen werden.[49] In einer großen Vergleichsstudie zur Vorhersage des Nervenzellverlustes anhand der Daten des Ventrikelvolumens und des Liquorvolumens über der Hirnoberfläche konnte gezeigt werden, dass ein vergrößertes Ventrikelvolumen im Falle der Schizophrenie mit einem reduzierten Volumen der grauen Substanz einhergehen könnte. Dies ist bemerkenswert, da ein vergrößertes Ventrikelvolumen üblicherweise auf das Konto einer Volumenreduktion der Basalganglien geht, was aber für die Schizophrenie nicht zutrifft.[50] In einer methodisch aufwendigen MRT-Studie konnte gezeigt werden, dass keine Unterschiede zwischen ersterkrankten und chronisch kranken Patienten mit einer Schizophrenie in Bezug auf ein vergrößertes Ventrikelvolumen bestehen, was mit der Hypothese einer neuronalen Entwicklungsstörung vereinbar ist.[51]
- Corticale Strukturen
- Temporallappen
Von allen corticalen Strukturen sind im Falle der Schizophrenie Veränderungen im Bereich des Temporallappens am besten untersucht.[52] Der sicherste Befund ist eine Volumenreduktion in dem Bereich des linken Schläfenlappens, in dem sich das Wernicke-Sprachzentrum befindet (Planum temporale/Gyrus temporalis superior). Diese Befunde konnten durch bildgebende Verfahren[53][54] und post-mortem Studien[55] bestätigt werden. Die Veränderungen korrelieren mit psychopathologischen Befunden.[56] Der mediale Abschnitt des Temporallappens ist Gegenstand besonderen Interesses, da sich hier Regionen befinden, in denen bei post-mortem-Studien Auffälligkeiten gefunden wurden. Im Bereich des enthorinalen Cortex haben Jakob und Beckmann bereits 1986 abnorme Neuronenpopulationen gefunden[57] und im Bereich des Hippocampus wurden in der Mehrzahl der durchgeführten Studien eine Volumenreduktion gefunden.[58] Außerdem zeigte sich in den post-mortem-Studien eine verminderte Neuronengröße, was auf eine gestörte Konnektivität der betroffenen Neuronen hinweist.[59][60]
- Frontallappen
Die am besten untersuchten Befunde bezüglich der Frontallappen bei Schizophrenie betreffen die metabolische Hypofrontalität.[61][62] Bezüglich morphologischer Veränderung ist bemerkenswert, dass die normale Asymmetrie des Frontallappens (rechts > links) bei Schizophrenie aufgehoben ist.[63] Da der Frontallappen in viele Untereinheiten unterteilt ist, sind Studien zu einzelnen Abschnitten dieser Hirnregion bei Schizophrenie durchgeführt worden. Dabei zeigte sich, dass es insbesondere im Bereich des dorsolateralen präfrontalen Cortex bei Schizophrenie eine Volumenminderung gibt.[64] In einzelnen Studien wurde in diesem Bereich auch eine Störung der Gyrifizierung festgestellt.[65]
- Parietallappen
Studien zu den Scheitellappen bei Schizophrenie ergaben bislang keine charakteristischen Veränderungen. In einzelnen Subregionen wurde eine Abnahme der grauen Substanz gefunden.[53]
- Subcorticale Strukturen
- Basalganglien
Die sogenannten Basalganglien sind der Ort von neuronalen Verbindungen zwischen verschiedenen Cortexarealen (parietal und präfrontal) und sind Teil einer sog. neuronalen Schleife vom Cortex über BG und Thalamus zurück zum Cortex. Diese Verbindungsstrukturen besorgen Auswahl und Veranlassung willentlicher Aktionen.[66] In 14 Studien in der Zeit von 1994 bis 2000 wurden in 80 % der Fälle strukturelle Veränderungen gefunden: Bei Patienten mit einer Schizophrenie, die mit Neuroleptika behandelt wurden zeigte sich eine Volumenzunahme der BG und bei neuroleptikanaiven Patienten eine Volumenminderung.[67][68]
- Thalamus
Der Thalamus gilt als eine Art Filter für sensorische Informationen zwischen Cortex und limbischen System.[69] In der Mehrzahl der durchgeführten MRT-Studien zeigte sich eine Volumenreduktion des Thalamus bei Schizophrenie. Dabei wurde einerseits gezeigt, dass die Volumenreduktion bilateral ist[70] und dass sie mit einer Minderperfusion einhergeht.[71]
- Cerebellum
Das Kleinhirn übt Kontrollfunktionen der Feinmotorik aus. Es gibt auch Hinweise für seine Beteiligung bei kognitiven Prozessen. Von Nancy Andreasen stammt die Theorie der „kognitiven Dysmetrie“. Sie besagt, dass das Kleinhirn über Verbindungen mit präfrontalem Cortex und Thalamus an den Symptomen der Schizophrenie beteiligt ist.[72] Die bisher durchgeführten Untersuchungen gründen sich auf den Befund einer Volumenminderung des Kleinhirnwurms.[73] Allerdings ist die Datenlage zu diesem Befund bislang eher schmal.
PET- und SPECT-Untersuchungen
Der bekannteste Befund aus funktionellen bildgebenden Untersuchungen mittels PET und SPECT ist das 1971 entdeckte Phänomen der Hypofrontalität bei schizophrenen Patienten. Franzen und Mitarbeiter beobachteten in ihrer Pionier-Studie eine Minderung der frontalen Hirndurchblutung.[74] Die frontale Minderperfusion beträgt bei allen seither durchgeführten Studien 1–8 %. Das Phänomen ist schon sehr bald mit der Vermutung verknüpft worden, dass bei Schizophrenen der frontale Cortex eine dopaminerge Minderaktivierung zeigt. Es ist anzunehmen, dass eine Art Gegenregulationsbemühung der frontalen Neurone zu einer dopaminergen Übersteuerung führt. Der durch die Hypofrontalität verursachte relative Dopaminüberschuss im limbischen System und in anderen corticalen Regionen könnte dann zu den psychotischen Symptomen der Schizophrenie führen.
FMRT- und MRS-Studien
Auch mithilfe der funktionellen Kernspintomographie wurde die Minderaktivierung des dorsolateralen präfrontalen Cortex bestätigt.[75] Besonders eindrucksvoll war der Nachweis, dass Patienten, die Stimmen hören, eine Aktivierung im primären akustischen Cortex zeigen.[76][77] Mithilfe der Phosphor-31-Magnet-Resonanz-Spektroskopie wurde ebenfalls ein verminderter Energieumsatz im Frontalhirn von schizophrenen Patienten gefunden. Mittels Wasserstoff-Spektroskopie kann man die neuronenspezifischen Substanzen (N-Acetyl-Aspartat) und den Marker für degenerative Prozesse Cholin nachweisen. Bei schizophrenen Patienten konnte übereinstimmend im Hippocampus eine Reduktion des NAA gefunden werden, bei unveränderten Werten für Cholin. Das bedeutet, dass die Minderaktivierung des Hippocampus bei Menschen mit einer Schizophrenie nicht auf degenerative Prozesse zurückzuführen ist. Zur Übersicht:[78]
Bildgebung und kognitive Leistungen
Die folgende Tabelle gibt einen Überblick über die Befunde zur Lokalisation von kognitiven Defiziten bei Patienten mit einer Schizophrenie.
| Kognitive Domäne | Beschreibung der kognitiven Leistung |
Hirnregion mit gestörter Signaländerung |
|---|---|---|
|
|
|
Neuropathologische Befunde
Aufgrund der radiologischen Befunde zur Ventrikelasymmetrie durch Huber und Johnstone kam es Anfang der 1980er-Jahre zu einer Renaissance der neuropathologischen Untersuchungen der Schizophrenien.
Limbisches System
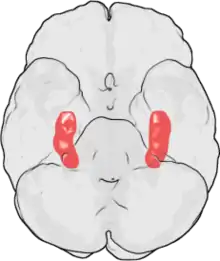
Den Beginn der modernen neuropathologischen Untersuchungen zur Schizophrenie bilden die Arbeiten von Bogerts.[79] Dessen Arbeitsgruppe hat ab 1984 in verschiedene Untersuchungen gezeigt, dass es bei Menschen mit einer Schizophrenie eine Volumenminderung im Bereich des Hippocampus und der Amygdala gibt. Volumenminderungen im Bereich des Thalamus sind gleichfalls beobachtet worden, die Befunde sind aber nicht so signifikant. Veränderungen sind auch im Bereich des Gyrus cinguli beschrieben worden.
Area entorhinalis
Die Area entorhinalis ist bei schizophrenen Patienten intensiv untersucht worden und die Befunde sind umstritten. Die Arbeitsgruppe um Jakob und Beckmann hat die Befunde gut dokumentiert,[80] andere konnten sie nicht reproduzieren. Zu einer Übersicht vgl.[81] Im Einzelnen handelt es sich um die Beobachtung von abnorm und rotiert gelagerten Neuronen. Das Hauptproblem dieser Untersuchungen ist die geringe Fallzahl: Insgesamt wurden bislang bei kaum mehr als zwei Dutzend Patienten post-mortem Studien zu diesem Zweck durchgeführt.
Migrationsstörungen
Ähnliche Veränderungen wie in der Area entorhinalis wurden teilweise auch im Hippocampus gefunden. Man vermutet, dass die Ursache dieser rotiert gelagerten Neurone Migrationsstörungen sind. Da man solche Veränderungen auch bei schizophrenen Opfern der Atombombenabwürfe in Hiroshima und Nagasaki fand, wurden Studien mit der Fragestellung durchgeführt, ob Menschen mit einer Schizophrenie gehäuft Mutationen im Bereich der DNA-Reparatursysteme haben. Diese Studien haben kein positives Ergebnis erbracht. Zu der Annahme einer genetisch bedingten oder früh erworbenen Migrationsstörung passen allerdings Beobachtungen, dass später schizophren Erkrankte als Kinder überdurchschnittlich häufig neurologische Defizite zeigten (motorische Ungeschicklichkeit oder atavistische Reflexe wie das Fingerspreizphänomen).
Volumenminderung
Neuroradiologische Untersuchungen haben bewiesen, dass es morphologische Veränderungen bei Patienten mit einer Schizophrenie gibt. Die Ursache dieser Veränderung wird seit etwa 20 Jahren intensiv erforscht. Die Befunde sind allerdings bislang nicht eindeutig interpretierbar. Eine Ursache dafür ist die geringe Zahl der Studien. Eine Übersicht von 2002 listet lediglich 70 post-mortem Untersuchungen auf. Nur sehr wenige Befunde sind eindeutig repliziert, nur wenige Studien sind methodisch vergleichbar. Obwohl beispielsweise die Volumenreduktion von Hippocampus und Amygdala bei schizophrenen Patienten durch bildgebende Verfahren inzwischen unbezweifelbar belegt ist, gibt es keine replizierten neuropathologischen Befunde zur Volumenreduktion oder Nervenzellverluste des Hippocampus. Die Ursache dieser Veränderung ist also bislang unklar.
Ursachen
Wiewohl die Datenlage für die neuropathologischen Untersuchungen unübersichtlich erscheint, gibt es doch eindeutige Befunde. Bei keiner der Untersuchungen wurden Hinweise für Veränderungen gefunden wie sie bei klassischen degenerativen Hirnprozessen zu erwarten sind. Die morphologischen Veränderungen bei der Schizophrenie sind also nicht mit denen bei Morbus Alzheimer, Multiple Sklerose oder Chorea Huntington vergleichbar. Zur Übersicht vgl.[82][83]
Sonstige organische Faktoren
Geburtskomplikationen
Mednick und Schulsinger haben Anfang der 1960er-Jahre behauptet, dass Geburtskomplikationen ein Risikofaktor für Schizophrenie seien.[84] Die bislang angestellten Untersuchungen ergaben aber, dass das Erkrankungsrisiko durch Geburtskomplikationen um maximal 1 % steigt. Gelegentlich wurde die Vermutung geäußert, dass umgekehrt die genetische Disposition zur Schizophrenie mit Reifungsstörungen einhergeht und dann sekundär möglicherweise zu Geburtskomplikationen führt.[85]
Infektions- und Immunhypothesen
Erste Infektions- und Immunhypothesen gehen auf Wagner-Jauregg zurück. Dieser erhielt den Nobelpreis für Medizin für seine Malaria-Experimente (Impfmalaria) bei psychotischen Patienten mit einer Lues. Es gibt keine soliden Daten, die diese Hypothesen belegen. Weder sind bei schizophrenen Patienten im Rahmen von postmortalen Untersuchungen Hinweise für das Vorliegen einer entzündlichen Erkrankung des ZNS gefunden worden, noch gibt es konsistente Ergebnisse bei Untersuchungen auf spezifische Antikörper gegen neurotrope Viren oder fremder DNA. Natürlich schließen die negativen Befunde nicht aus, dass Infektionen mit Erregern vorliegen, die sich ins Genom integrieren, oder die sich nur intrazellulär vermehren (Borrelien). Es gibt allerdings epidemiologische Daten, die für die Infektionshypothese sprechen: Retrospektiv fand man, dass das Erkranken der Mutter eines späteren Patienten an einer Virus-Grippe während Grippeepidemien zu der Zeit des zweiten Trimenon das Risiko des Ungeborenen, später an einer Schizophrenie zu erkranken, erhöhen. Auch gibt es ein Überwiegen der Wintergeburten von schizophrenen Patienten auf der Nordhalbkugel, was mit einem erhöhten Erkrankungsrisiko der Mutter in der Zeit des zweiten Trimenon für Virusinfektionen einhergeht.
Neuere Forschungen weisen in Richtung einer unspezifischen Aktivierung bestimmter Teile des Immunsystems.[86] Auch Hinweise Veränderungen des Tryptophan- und Kynureninstoffwechsels verdichten sich. Störungen des Kynureninstoffwechsels beim Menschen mit erhöhten L-Kynurenin bzw. Metaboliten-Werten sind für die Schizophrenie[87][88] und andere psychiatrische Erkrankungen[89] beschrieben. Typischerweise kommt es dabei zu einer Anhäufung (Kumulation) von Kynurenin und einer Verschiebung des Tryptophanstoffwechsels hin zu Kynurensäure, Anthranilsäure und deren weiteren Stoffwechselprodukten.[87][90][89][91][92][93] Eine häufige Konstellation bei verschiedenen neuropsychiatrischen Erkrankungen ist eine gleichzeitig erhöhte Kynurenin/Tryptophan ratio durch Akkumulation von Kynurenin vor dem nächsten Stoffwechselschritt, der Hydroxylierung zu 3-Hydroxykynurenin infolge Katalysierung durch Kynurenin-3-Monooxygenase (KMO).[94][95][88]
Literatur
- Matcheri S. Keshavan, Rajiv Tandon, Nash N. Boutros, Henry A. Nasrallah: Schizophrenia. “just the facts”: What we know in 2008. Part 3: Neurobiology. In: Schizophrenia Research, 106 (2008), S. 89–107.
- Christopher A. Ross, Russell L. Margolis, Sarah A.J. Reading, Mikhail Pletnikov, Joseph T. Coyle: Neurobiology of Schizophrenia. In: Neuron, 52 (2006), S. 139–153.
Einzelnachweise
- J. Kornhuber, J. Wiltfang, S. Bleich: The aetiopathogenesis of schizophrenias. In: Pharmacopsychiatry, 37 (2004), Suppl. 2, S. 103–112.
- Hans-Walter Schmuhl (Hrsg.): Rassenforschung an Kaiser-Wilhelm-Instituten vor und nach 1933. Wallstein-Verlag, Göttingen 2003, ISBN 3-89244-471-4.
- Volker Roelcke: Programm und Praxis der psychiatrischen Genetik an der Deutschen Forschungsanstalt für Psychiatrie unter Ernst Rüdin. In: Schmuhl: Rassenforschung.
- Ernst Rüdin: Zur Vererbung und Neuentstehung der Dementia praecox. Berlin 1916.
- E. Salter, V. Cowie (Hrsg.): Genetics of Mental Disorders. Oxford 1970.
- M. Fischer u. a. In: Br. J. Psychiatry, 115 (1969), S. 981–990, PMID 5387002
- S. Onstad u. a. In: Acta Psychiatr. Scand., 83 (1991), S. 395–401, PMID 1853734
- Seymour S. Kety u. a.: Mental illness in the biological and adoptive relatives of schizophrenic adoptees. Replication of the Copenhagen Study in the rest of Denmark. In: Arch Gen Psychiatry 51 (1994), S. 442–455, PMID 8192547
- I. I. Gottesman, A. Shields: A critical review of recent adoption, twin and family studies of schizophrenia: behavioral genetics perspectives. In: Schizophrenia Bulletin, 2(1976), S. 360–401, PMID 1034336.
- C. M. Lewis u. a.: Genom-scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. In: American Journal of human genetics, 2003, 73, 1, S. 34–48, PMID 12802786.
- G. Kirov u. a.: Finding Schizophrenia Genes. In: J. Clini. Invest., 2005 115 (6), S. 1440–1448, PMID 15931379.
- A. S. Bassett: Chromosomal aberrations and schizophrenia. Autosomes. In: British Journal of Psychiatry, 161 (1992), S. 323–334, PMID 1393302.
- G. Silberberg, A. Darvasi, R. Pinkas-Kramarski, R. Navon: The involvement of ErbB4 with schizophrenia: association and expression studies. In: Am J Med Genet B Neuropsychiatr Genet., 2006 Mar 5; 141(2), S. 142–148, PMID 16402353
- D. R. Weinberger: Genetic mechanisms of psychosis: in vivo and postmortem genomics. In: Clin Ther., 2005; 27 Suppl A: S8 – 15, PMID 16198200
- N. Norton, H. J. Williams, M. J. Owen: An update on the genetics of schizophrenia. In: Curr Opin Psychiatry, 2006 Mar; 19(2), S. 158–164, PMID 16612196
- George Kirov, Michael C. O’Donovan, Michael J. Owen: Finding schizophrenia genes. In: J Clin Invest., 2005 June 1; 115(6), S. 1440–1448, PMID 15931379
- M. J. Owen, N. Craddock, M. C. O’Donovan: Schizophrenia: genes at last? In: Trends Genet. 2005 Sep; 21(9), S. 518–525, PMID 16009449
- P. J. Harrison, D. R. Weinberger: Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. In: Mol Psychiatry, 2005 Jan; 10(1), S. 40–68, PMID 15263907
- B. H. Shirts, V. Nimgaonkar: The genes for schizophrenia: finally a breakthrough? In: Curr Psychiatry Rep., 2004 Aug; 6(4), S. 303–312, PMID 15260947
- S. Mah u. a.: Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. In: Mol Psychiatry, (2006) 11, S. 471–478, PMID 16402134.
- H. Häfner (Hrsg.): Risk and protective factors in schizophrenia. Towards a conceptual model of the disease process. Steinkopff, Darmstadt 2002.
- P. Propping: Psychiatrische Genetik. Befunde und Konzepte. Springer, Berlin 1989.
- N. L. Ross, R. Wadekar, A. Lopes, A. Dagnall, J. Close, L. E. Delisi, T. J. Crow: Methylation of two Homo sapiens-specific X-Y homologous genes in Klinefelter’s syndrome (XXY). In: Am J Med Genet B Neuropsychiatr Genet., 2006 Jul 5; 141(5), S. 544–548, PMID 16741946
- A. Carlson: Antipsychotic drugs, neurotransmitters and schizophrenia. In: American Journal of Psychiatry, 135, 1978, S. 164–173.
- S. H. Snyder: The dopamine hypothesis of schizophrenia: focus on the dopamine receptor. In: Am J Psychiatry. 1976 Feb; 133(2), S. 197–202, PMID 1251927
- A. S. Horn, S. H. Snyder: Chlorpromazine and dopamine: conformational similarities that correlate with the antischizophrenic activity of phenothiazine drugs. In: Proc Natl Acad Sci U S A, 1971 Oct, 68(10), S. 2325–2328, PMID 5289865.
- S. H. Snyder: Catecholamines in the brain as mediators of amphetamine psychosis. In: Arch Gen Psychiatry, 1972 Aug; 27(2), S. 169–179, PMID 4339577.
- S. H. Snyder: Amphetamine psychosis: a “model” schizophrenia mediated by catecholamines. In: Am J Psychiatry., 1973 Jan, 130(1), S. 61–67, PMID 4345465.
- T.J. Crow: Molecular pathology of schizophrenia: more than one disease process? In: Br Med J, 280, 1980, S. 66–68, PMID 6101544.
- N. Yu, K. R. Tucker, E. S. Levitan, P. D. Shepard, C. C. Canavier: Implications of cellular models of dopamine neurons for schizophrenia. In: Progress in molecular biology and translational science. Band 123, 2014, S. 53–82, doi:10.1016/B978-0-12-397897-4.00011-5, PMID 24560140, PMC 4351765 (freier Volltext) (Review).
- RD Paz, S Tardito, M Atzori, KY. Tseng: Glutamatergic dysfunction in schizophrenia: from basic neuroscience to clinical psychopharmacology. In: Eur Neuropsychopharmacol. 2008 Nov; 18(11): 773-86. Epub 2008 Jul 23. Review, PMID 18650071
- EH Wong, JA Kemp, T Priestley, AR Knight, GN Woodruff, LL. Iversen: The anticonvulsant MK-801 is a potent N-methyl-D-aspartate antagonist. In: Proc Natl Acad Sci U S A. 1986 Sep; 83(18), S. 7104–7108, PMID 3529096
- D.C. Goff, J.T. Coyle: The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. In: American Journal of Psychiatry, 158, 2001, S. 1367–1377, PMID 11532718.
- D.E. Nichols: Hallucinogens. In: Pharmacol Ther, 101, 2004, S. 131–181, PMID 14761703
- N.J. Penington u. a.: Effects of LSD on Calcium-currents in central 5-HT-containing neurons: 5-HT(1A) receptors may play a role in hallucinogenesis. In: J. Pharmacol. Exp. Ther., 269, 1994, S. 1160–1165, PMID 8014859.
- H. Osmond, J. Smythies: Schizophrenia: new approach. In: J. Mental. Sci., 98, 1952, S. 309–322, PMID 14917992.
- M Wang u. a.: Schizophrenia, amphetamine-induced sensitized state and acute amphetamine exposure all show a common alteration: increased dopamine D2 receptor dimerization. In: Mol Brain, 2010, 3, S. 25, PMID 20813060
- A. Alzheimer: Beiträge zur pathologischen Anatomie der Hirnrinde und zur anatomischen Grundlage einiger Psychosen. In: Mschr. Psychiat. Neurol. 2, S. 82–120, 1897.
- G Peters: Neuropathologie und Psychiatrie. In: H.W. Gruhle, (Hrsg.): Psychiatrie der Gegenwart. Springer, Berlin 1967, Band I, S. 298–305.
- G. Huber: Chronische Schizophrenie. Synopsis klinischer und radiologischer Untersuchungen an defekt-schizophrenen Anstaltspatienten. Hüthig, Heidelberg, 1961.
- Gergely Klinda: Zur Geschichte der Pneumenzephalographie. Dissertation, Charité Berlin 2010
- E.C. Johnstone u. a.: Cerebral ventricular size and cognitive impairement in chronic schizophrenia. In: Lancet, 2, 1976, S. 924–926, PMID 62160.
- J.D. Van Horn u. a.: Ventricular enlargement in schizophrenia. A meta-analysis of studies of the ventricle: brainratio (VBR). In: Br. J. Psychiatry, 160, 1992, S. 687–697, PMID 1534268.
- A. Schmitt u. a.: Aktueller Überblick über strukturelle Magnetresonanztomographie bei Schizophrenie. In: Fortschritte Neurologie Psychiatrie, 2001, 69, S. 105–115, PMID 11305121
- RE Gur u. a.: Reduced grey matter volume in schizophrenia. In: Arch Gen Psychiatry, 1999, 56, S. 905–911, PMID 10530632
- RB Zipursky u. a.: Cerebral grey matter volume deficits in first episode psychosis. In: Arch Gen Psychiatry, 1998, 55, S. 540–546, PMID 9633673
- C. Arango u. a.: The relationship of clozapin and haloperidol treatment response to prefrontal, hippocampal and caudate brain volumes. In: Neuroreport, 2003, 14, S. 2025–2029, PMID 12900303
- DF Braus: Schizophrenie. Schattauer Verlag, 2005, ISBN 3-7945-2316-4, S. 116.
- PJ. Harrison: Brains at risk of schizophrenia. In: Lancet, 1999, 353, S. 3–4, PMID 10023939
- LL Symonds u. a.: Does an increase in sulcal or ventricular fluid predict where brain tissue is lost? In: J Neuroimaging, 1999, 9, S. 201–209, PMID 10540599.
- DF Braus: Schizophrenie. Schattauer Verlag, 2005, ISBN 3-7945-2316-4, S. 118 f.
- IC Wright u. a.: Meta-analysis of regional brain volumes in schizophrenia. In: Am J Psychiatry, 2000, 157, S. 16–25, PMID 10618008
- RW. McCarley u. a.: MRI anatomy of schizophrenia. In: Biol Psychiatry, 1999, 45, S. 1099–1119, PMID 10331102
- RP Rajarethinam u. a.: Superior temporal gyrus in schizophrenia: a volumetric MRI study. In: Schizophrenia Research, 2000; 41, S. 303–312, PMID 10708339
- P. Falkai u. a.: Disturbed planum temporale asymmetry in schizophrenia. In: Schizophrenia Research, 1995; 14, S. 161–176, PMID 7710997
- C. Levitan u. a.: Superior temporal gyral volumes and laterality correlates of auditory hallucinations in schizophrenia. In: Biol Psychiatry, 1999, 46, S. 955–962, PMID 10509178
- H Jakob, H. Beckmann: Prenatal developmental disturbances in the limbic allocortex in schizophrenics. In: J Neural Transm, 1986, 65, S. 303–326, PMID 3711886
- SE Arnold u. a.: Smaller neuron size in schizophrenia in hippocampal subfields that mediate cortical-hippocampal interactions. In: Am J Psychiatry, 1995, 152, S. 738–748, PMID 7726314
- K Henke u. a.: Human hippocampus associates information in memory. In: PNAS, 1999; 96, S. 5884–5889, PMID 10318979
- DW Zaidel u. a.: Size, shape and orientation of neurons in the left and right hippocampal investigation of normal asymmetries and alterations in schizophrenia. In: Am J Psychiatry, 1997, PMID 9167509
- CS Carter u. a.: Functional hypofrontality and working memory dysfunction in schizophrenia. In: Am J Psychiatry, 1998; 155, S. 1285–1287, PMID 9734557
- J Schröder u. a.: Cerebral metabolic activity correlates of subsyndroms in chronic schizophrenia. In: Schizophrenia Research, 1996; 19, S. 41–53, PMID 9147495
- RM Bilder u. a.: Absence of regional hemispheric volume asymmetries in first episode schizophrenia. In: Am J Psychiatry, 1994; 151, S. 1437–1447, PMID 8092337
- TE Schlaepfer u. a.: Decreased regional cortical grey matter volume in schizophrenia. In: Am J Psychiatry, 1994; 151, S. 842–848, PMID 8184992
- K Vogeley u. a.: Right frontal hypergyria differentiation in affected and unaffected siblings from families multiply affected with schizophrenia: a morphometric MRI study. In: Am J Psychiatry, 2001; 158, S. 494–496, PMID 11229998
- S. Heckers: Neuropathology of Schizophrenia. In: Schizophrenia Bulletin, 1997, 23, S. 403–421, PMID 9327506
- MH Chakos u. a.: Caudate nuclei volumes in schizophrenic patients treated with typical antipsychotics or clozapine. In: Lancet, 1995; 345, S. 356–357, PMID 7853978
- AM Elkashef u. a.: Basal ganglia pathology in schizophrenia and tardive dyskinesia: an MRI quantitative study. In: Am J Psychiatry, 1995; 151, S. 752–755, PMID 7909412
- A Carlsson u. a.: Neurotransmitter aberrations in Schizophrenia. In: Life Sci, 1997; 61, S. 75–94, PMID 9217267
- C Gaser u. a.: Detecting structural changes in whole brain based on nonlinear deformations-application to schizophrenia research. In: Neuroimage, 1999; 10, S. 107–113, PMID 10417245
- MS Buchsbaum u. a.: PET and MRI of the thalamus in never – medicated patients with schizophrenia. In: Am J Psychiatry, 1996; 153, S. 191–199, PMID 8561198
- NC Andreasen u. a.: Defining the phenotype of schizophrenia: cognitive dysmetria and ist neural mechanisms. In: Biol Psychiatry, 1999; 46, S. 908–920, PMID 10509174
- G Ende u. a.: Further evidence for altered cerebellar neuronal integrity in schizophrenia. In: Am J Psychiatry, 2005; 162, S. 790–792, PMID 15800155
- G. Franzen, D.H. Ingvar: Absence of activation in frontal structures during psychological testing of chronic schizophrenics. In: J Neurol Neurosurg Psychiatry, 38 (1975) S. 1027–1032, PMID 1202164.
- D.R. Weinberger u. a.: fMRI applications in schizophrenia research. In: Neuroimage, 4, 1996, S. 118–126, PMID 9345536.
- T. Dierks u. a.: Activation of Heschls gyrus during auditory hallucinations. In: Neuron, 22, 1999, S. 615–621, PMID 10197540.
- P.K. McGuire u. a.: Increased blood flow in Brocas Area during auditory hallicinations in schizophrenia. In: Lancet, 342, 1993, S. 703–705, PMID 8103821.
- T. Kircher u. a.: Funktionelle Bildgebung am Beispiel der Schizophrenie. In: Deutsches Ärzteblatt, 101, 2004, S. A1975–1980.
- B. Bogerts: Neuropathologie der Schizophrenien. In: Fortschr. Neurol. Psychiat, 52, S. 428–437, 1984, PMID 6519627
- H. Jakob, H. Beckmann: Prenatal developmental disturbances in the limbic allocortex in schizophrenics. In: J. Neural Transm., 1986, 65, 303, PMID 3711886
- P. Falkai u. a.: Entorhinal cortex pre-alpha cell-clusters in schizophrenia: quantitative evidence of a developmental abnormality. In: Biol Psychiatry, 47, 2000, S. 937–943, PMID 10838061
- P. Falkai u. a.: Neuropathology. In: M.G. Gelder (Hrsg.): New Oxford Textbokk of Psychiatry. 2000.
- P. Falkai u. a.: No evidence for astrogliosis in brains of schizophrenic patients. A post-mortem study. In: Neuropathol Appl Neurobiol. 25, S. 48–53, 1999, PMID 10194775
- S.A. Mednick, F. Schulsinger: Studies of children at high risk for schizophrenia, S. 247–293. In: S.R. Dean (Hrsg.): Schizophrenia. MSS Information, New York 1973, ISBN 0-8422-7115-5.
- T.F. McNeil: Review Article. Obstetric complications in schizophrenic parents. In: Schizophr. Res., 5 (1991) S. 89–101, PMID 1931811.
- Norbert Müller, Markus J. Schwarz: Immune System and Schizophrenia. In: Curr Immunol Rev., 2010, 6, S. 213–220.
- Ikwunga Wonodi, O. Colin Stine, Korrapati V. Sathyasaikumar et al.: Downregulated Kynurenine 3-Monooxygenase Gene Expression and Enzyme Activity in Schizophrenia and Genetic Association With Schizophrenia Endophenotypes. In: Arch Gen Psychiatry. 2011; 68, S. 665–674.
- Maria Holtze, Peter Saetre, Göran Engberg, et al.: Kynurenine 3-monooxygenase polymorphisms: relevance for kynurenic acid synthesis in patients with schizophrenia and healthy controls. In: J Psychiatry Neurosci. 2012; 37, S. 53–57.
- PJ Hoekstra, GM Anderson, PW Troost: Plasma kynurenine and related measures in tic disorder patients. In: Eur Child Adolesc Psychiatry. 2007; 16 Suppl 1, S. 71–77.
- Brian M. Campbell, Erik Charych,Anna W. Lee, Thomas Möller: Kynurenines in CNS disease: regulation byinflammatory cytokines. In: Frontiers in Neuroscience. Neuroendocrine Science 2014, Volume 8, Article 12.
- Serdar M. Dursun, Gillian Farrar, Sheila L. Handley, et al.: Elevated plasma kynurenine in Tourette syndrome. In: Molecular and Chemical Neuropathology, 1994, 21, S. 55–60
- H. Rickards, S. M. Dursuna, G. Farrar: Increased plasma kynurenine and its relationship to neopterin and tryptophan in Tourette’s syndrome. In: Psychological Medicine, 26, 1996, S. 857–862
- Erik Kwidzinski: Beteiligung der Indolamin 2, 3-Dioxygenase (IDO) an Immunregulation des zentralen Nervensystems. Dissertation, Charité Berlin, 13. Februar 2006, urn:nbn:de:kobv:11-10059777
- K. Schroecksnadel, S. Kaser, G. Neurauter, et al.: Increased Degradation of Tryptophan in Blood of Patients with Rheumatoid Arthritis. In: The Journal of Rheumatology 2003; 30: 9
- M Maes, R Verkerk, S Bonaccorso et al.: Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation. In: Life Sci. 2002 Sep 6; 71(16): 1837-48.