Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel
Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel, auch MCAD-Mangel (engl. medium-chain acyl-CoA dehydrogenase deficiency (MCADD)) genannt, ist eine der häufigsten angeborenen Stoffwechselerkrankungen. Es handelt sich um eine Proteinfehlfaltungserkrankung mit loss of function bei der durch eine Mutation das Enzym Medium-Chain-Acyl-CoA-Dehydrogenase (MCAD) in seiner Funktion beeinträchtigt ist.[1][2] Der Defekt in MCAD bewirkt einen verminderten Abbau mittelkettiger (medium chain) Fettsäuren. Diese Fettsäuren können deshalb im Intermediärstoffwechsel nur mangelhaft genutzt werden.[3] Der Erbgang des MCAD-Mangels ist autosomal-rezessiv.
| Klassifikation nach ICD-10 | |
|---|---|
| E71.3 | Störungen des Fettsäurestoffwechsels |
| ICD-10 online (WHO-Version 2019) | |
Pathologie
Zur energetischen Nutzung in den Zellen werden Fettsäuren schrittweise durch Entfernung kleiner Moleküle um jeweils zwei Kohlenstoffatome verkürzt. Unter normalen Bedingungen werden die Fettsäuren unter Freisetzung von Energie in Kohlendioxid und Wasser umgewandelt. Dieser Prozess wird β-Oxidation genannt. Ein Komplex von sieben Enzymen ist nötig um jede Zweier-Einheit zu entfernen. Da der Prozess das Entfernen von Wasserstoffatomen aus einer Acylgruppe beinhaltet, wird der Enzymkomplex Acyl-Dehydrogenase genannt. Der Oxidationsprozess der Fettsäuren geschieht innerhalb der Mitochondrien. Fettsäuren des Zytoplasmas werden zum Transport durch die mitochondriale Membran an ein Carnitin-Molekül gebunden. Die Kombination aus Carnitin und Fettsäure wird Acyl-Carnitin genannt. Patienten mit MCAD-Mangel haben eine erhöhte Konzentration von mittelkettigen Acyl-Carnitinen im Zytoplasma der Zellen, da sie die Acyl-Carnitine nur unzureichend abbauen können. Die mittelkettigen Fettsäuren bestehen aus 6 bis 12 Kohlenstoffatomen. Die mittelkettigen Acyl-Carnitine werden zum Teil von den Zellen sezerniert und gelangen so in den Blutkreislauf.
Patienten mit verringerter MCAD-Aktivität haben eine Störung der Fettsäureoxidation. Bei normaler Gesundheit führt dies zu keinen Beeinträchtigungen. Die Störung kann aber im Falle einer weiteren Krankheit, welche eine längere Fastenperiode verursacht, zu Hypoglykämie (Unterzuckerung), Hyperammonämie und möglicherweise plötzlichem Tod führen. Bei frühzeitiger Entdeckung des MCAD-Mangels kann in diesen Situationen einer Stoffwechselentgleisung aber durch ausreichende Nahrungszufuhr und notfalls durch Glucoseinfusionen im Krankenhaus sehr sicher entgegengewirkt werden. Während bis vor wenigen Jahren ein unentdeckter MCAD-Mangel beim ersten Auftreten einer Stoffwechselentgleisung noch in etwa 25 % der Fälle zum Tode führte, lässt sich das Eintreten einer solchen metabolischen Krise, aufgrund der inzwischen frühzeitig erfolgenden Feststellung eines MCAD-Mangel, in fast allen Fällen sehr wirkungsvoll verhindern.
Untersuchung bei plötzlichem Säuglingstod
In einigen wenigen Fällen (ca. 1 %) des plötzlichen Säuglingstodes konnte im Nachhinein ein MCAD-Mangel festgestellt werden. Aufgrund dieser Seltenheit wird zwar eine ursächliche Beteiligung vermutet, einen sicheren Beweis für den MCAD-Mangel als eigentliche Todesursache gibt es speziell im Hinblick auf den plötzlichen Säuglingstod jedoch nicht.
Bei der Untersuchung eines Kindes, das dem plötzlichen Säuglingstod erlag, werden Blutproben zur Bestimmung der Acyl-Carnitin-Konzentration des Blutes entnommen. Auf MCAD-Mangel kann geschlossen werden, wenn der Acyl-Carnitin-Spiegel des Blutes auf einen typischen Wert angestiegen ist.
Wenn die Zeitspanne zwischen plötzlichem Säuglingstod und der Obduktion nicht zu lange ist, ist es manchmal möglich, Fibroblasten der Dermis einer Hautprobe während der Untersuchung zu entnehmen. Es können mit radioaktiven Kohlenstoffatomen (C-14) markierte Fettsäuren in das kultivierte Medium gegeben werden. Wenn die Zellen diese Fettsäuren oxidieren und innerhalb ihres Stoffwechsels absorbieren, wird radioaktives Kohlendioxid erzeugt, das mit einem geeigneten Gerät nachgewiesen werden kann.
Die Produktionsrate von Kohlendioxid aufgrund von Fettsäureketten unterschiedlicher Länge, können als Test zur Bestimmung, ob ein Mangel einer der Acyl-Dehydrogenasen vorliegt, verwendet werden. Dieser Test kann zur Unterstützung der MCAD-Mangel-Diagnose benutzt werden, wenn der Verdacht aufgrund des Musters der Acyl-Carnitinen besteht.
Genetik
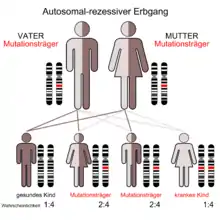
Mutationen im ACADM-Gen führen zu einer Fehlfaltung des MCAD-Enzyms. Das Gen befindet sich beim Menschen auf Chromosom 1 Genlocus p31. Die fehlgefalteten Enzyme werden von der Proteinqualitätskontrolle ausgesondert und im Proteasom zerlegt. Die betroffenen Patienten weisen deshalb einen Mangel an Medium-Chain-Acyl-CoA-Dehydrogenase auf. Die Erkrankung wird autosomal-rezessiv vererbt.[3] Dies bedeutet, dass zwei Kopien eines fehlerhaften Gens in jeder Zelle notwendig sind, um den Mangel herbeizuführen. Wenn nur eine Kopie fehlerhaft ist, dann ist die Person ein asymptomatischer Träger und erkrankt nicht an MCAD-Mangel. Man geht beim MCAD-Mangel von einer Häufigkeit von etwa 1:10.000 aus.[3][4][5]
Diagnose
Im Plasma lassen sich die mittelkettigen Acyl-Carnitine beispielsweise mittels der Tandem-Massenspektrometrie nachweisen. Dies kann im Rahmen eines Neugeborenen-Screenings geschehen. Eine erhöhte Konzentration von aktivierten Fettsäuren mittlerer Kettenlänge (6–12 Kohlenstoffeinheiten) innerhalb der Acyl-Carnitine legt den Verdacht auf einen MCAD-Mangel nahe. Als Leitparameter dient dabei die Konzentration von Octanoylcarnitin. Ein auffälliger Erstbefund wird üblicherweise durch ein Kontrollscreening und gegebenenfalls durch eine molekulargenetische Untersuchung (DNA-Analyse) überprüft.[3]
Der klinische Verdacht auf einen MCAD-Mangel kann durch ein Blutbild erhärtet werden, wenn gleichzeitig eine Hypoglykämie, Metabolische Azidose sowie Hyperurikämie diagnostiziert werden.[6][7]
Varianten
Vor der Einführung des erweiterten Neugeborenen-Screenings wurde der MCAD-Mangel in den meisten Fällen erst in den Untersuchungen nach einer akuten Stoffwechselentgleisung festgestellt. Dabei wurde in rund 80 % der Fälle die Mutation K329E (andere Bezeichnung c.985A>G; eine Punktmutation) homozygot, d. h. auf beiden Genkopien gefunden. In weiteren 18 % der Fälle war dieser Gendefekt in Verbindung mit einer anderen Mutation (compound heterozygot) beteiligt. Aus diesem Grund wird K329E weltweit als Hochrisikovariante eingestuft.
In den molekulargenetischen Untersuchungen nach auffälligen Screening-Befunden werden inzwischen jedoch auch eine ganze Reihe weiterer Mutationskombinationen gefunden, die nie zuvor klinisch in Erscheinung traten. Man nimmt daher an, dass es sich in den meisten dieser Fälle um einen milden Phänotyp des MCAD-Mangels handelt. Patienten mit diesen milden Varianten werden vermutlich auch ohne Behandlung zeitlebens symptomlos bleiben, jedoch ließe sich ein stichhaltiger Beweis nur mit einem entsprechend ausgedehnten Fastentest (> 24 Stunden) erbringen. Da ein solcher Test mit dem erhöhten Risiko einer dennoch eintretenden Stoffwechselentgleisung verbunden wäre, werden üblicherweise sowohl Patienten mit der Hochrisikovariante, als auch solche mit einer wahrscheinlich milden MCAD-Mangel-Ausprägung in den Stoffwechselzentren in gleicher Weise (z. B. mit Glucoseinfusionen im Krankheitsfall) behandelt.
Erstbeschreibung
Der Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel wurde erstmals 1976 von dem dänischen Molekulargenetiker Niels Gregersen und Kollegen beschrieben.[8]
Einzelnachweise
- LMU München: Proteinfehlfaltung.
- E. M. Maier, S. W. Gersting u. a.: Protein misfolding is the molecular mechanism underlying MCADD identified in newborn screening. In: Human molecular genetics. Band 18, Nummer 9, Mai 2009, S. 1612–1623, ISSN 1460-2083. doi:10.1093/hmg/ddp079. PMID 19224950. PMC 2667288 (freier Volltext).
- I. Knerr, U. Nennstiel-Ratzel u. a.: Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel: eine klinisch bedeutsame Stoffwechselstörung. In: Dtsch Arztebl. Band 102, 2005, S. A-2565/B-2166/C-2045.
- G. F. Hoffmann, R. von Kries u. a.: Frequencies of inherited organic acidurias and disorders of mitochondrial fatty acid transport and oxidation in Germany. In: European Journal of Pediatrics. Band 163, Nummer 2, Februar 2004, S. 76–80, ISSN 0340-6199. doi:10.1007/s00431-003-1246-3. PMID 14714182.
- A. Schulze, M. Lindner u. a.: Expanded newborn screening for inborn errors of metabolism by electrospray ionization-tandem mass spectrometry: results, outcome, and implications. In: Pediatrics. Band 111, Nummer 6 Pt 1, Juni 2003, S. 1399–1406, ISSN 1098-4275. PMID 12777559.
- Hyperurikämie als Leitsymptom beim Acyl-Coenzym A-Dehydrogenasedefekt mittelkettiger Fettsäuren. In: Klin Padiatr. 1997; 209(6), S. 357–360, doi:10.1055/s-2008-1043975.
- Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel: eine klinisch bedeutsame Stoffwechselstörung. In: Dtsch Arztebl. 2005; 102(38), S. A-2565 / B-2166 / C-2045.
- N. Gregersen, R. Lauritzen, K. Rasmussen: Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. In: Clinica chimica acta. Band 70, Nummer 3, August 1976, S. 417–425, ISSN 0009-8981. PMID 947635.
Weblinks
- Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel. In: Online Mendelian Inheritance in Man. (englisch)
- Medium-Chain-Acyl-CoA-Dehydrogenase-Mangel. In: Orphanet (Datenbank für seltene Krankheiten).
- Medium chain acyl-CoA dehydrogenase deficiency newbornscreening.info (englisch)
- ACADM gene (U.S. National Library of Medicine)