Joback-Methode
Die Joback-Methode[1] (oft auch als Joback-Reid-Methode bezeichnet) erlaubt die Vorhersage von elf wichtigen thermodynamischen Eigenschaften reiner Stoffe ausschließlich aus der Molekülstruktur.
Grundlagen
Gruppenbeitragsmethode
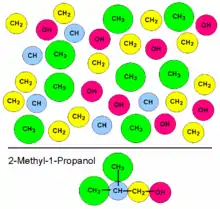
Die Joback-Methode ist eine Gruppenbeitragsmethode. Dieser Typ von Vorhersagemethoden verwendet einfache strukturelle Informationen eines chemischen Moleküls wie etwa einer Liste der funktionellen Gruppen, weist diesen Gruppen Parametern zu und berechnet thermophysikalische und Transporteigenschaften als eine Funktion der Summe dieser Gruppenparameter.
Joback nahm an, dass es keine Wechselwirkungen zwischen den Gruppen gibt und verwendete daher ausschließlich additive Gruppenbeiträge und keine Beiträge für Wechselwirkungen zwischen den Gruppen. Andere Methoden, wie beispielsweise UNIFAC, das Gemischeigenschaften wie etwa Aktivitätskoeffizienten abschätzt, oder die Benson-Methode, die Wärmekapazitäten, Bildungsenthalpien und -entropien abschätzt, verwenden neben den einfachen rein additiven Beiträge auch Wechselwirkungsparameter. Der große Vorteile der Beschränkung auf einfache Beiträge besteht in der geringen Anzahl der benötigten Parameter (ein Parameter für jede Gruppe und Eigenschaft), während die Anzahl der Wechselwirkungsparameter sehr stark mit steigender Anzahl von Gruppen ansteigt (1 für zwei Gruppen, 3 für drei, 6 für vier, 45 für zehn und sogar noch zweimal so viel, wenn die Wechselwirkungsparameter nicht symmetrisch sind).
Neun der vom Joback-Modell vorhergesagten Eigenschaften sind temperaturunabhängige Größen, die meisten werden schlicht aus der Summe der Gruppenbeiträge plus einem Summanden berechnet.
Zwei der Eigenschaften sind temperaturabhängig: Die Ideal-Gas-Wärmekapazität und die dynamische Viskosität von Flüssigkeiten. Für die Wärmekapazität wird ein kubisches Polynom mit vier Parametern verwendet, für die Flüssigviskosität ein Polynom mit lediglich zwei Parametern (Gerade). In beiden Fällen werden die Gleichungsparameter aus Gruppenbeträgen bestimmt.
Geschichte
Die Joback-Methode ist eine Erweiterung der Lydersen-Methode[2] und benutzt sehr ähnliche Gruppen, Formeln und Parameter für die Eigenschaften, die Lydersen auch bereits unterstützte (kritische Temperatur, kritischer Druck und kritisches Volumen).
Joback erweiterte das Modell für weitere Eigenschaften, bestimmte neue Parameter und modifizierte leicht die Bestimmungsgleichungen des alten Lydersen-Modells.
Modellstärken und -schwächen
Stärken
Die Popularität der Joback-Methode leitet sich im Wesentlichen von der einen Gruppenliste ab, die für alle Eigenschaften gleich ist. Dies erlaubt die Vorhersage aller elf unterstützten Eigenschaften aus einer einzigen Analyse einer chemischen Strukturformel.
Zusätzlich sind die Gruppen des Joback-Modells sehr einfach gehalten und können auch bei nur geringen chemischen Kenntnissen verwendet werden.
Schwächen
Neuere Entwicklungen von Abschätzverfahren[3][4] haben gezeigt, dass die Qualität der Joback-Methode begrenzt ist. Auch die Originalautoren haben bereits in ihrer Veröffentlichung festgestellt: „High accuracy is not claimed, but the proposed methods are often as or more accurate than techniques in common use today.“ (dt. etwa: „Hohe Genauigkeit wird nicht behauptet, jedoch sind die vorgeschlagenen Methoden oft so genau oder genauer als aktuell gebräuchliche Verfahren.“)
Die Liste der Gruppen deckt viele gebräuchliche Komponenten nur unzureichend ab. Insbesondere aromatische Stoffe werden nicht unterschieden von normalen Ringkomponenten. Dies ist ein ernsthaftes Problem, da sich die Eigenschaften dieser Komponentenklassen deutlich unterscheiden.
Die Datenbasis, die Joback und Reid zur Bestimmung der Gruppenparameter verwendeten, war recht klein und deckten nur eine geringe Anzahl verschiedenartiger Stoffe ab. Die beste Datenbasis wurde für Normalsiedepunkte erreicht (438 Komponenten) und die schlechteste für die Schmelzenthalpie (155 Komponenten). Die Vorhersage der Schmelzenthalpie ist darüber hinaus mit Gruppenbeitragsmethoden nur sehr schlecht möglich. Joback und Reid schreiben daher selbst, dass die Genauigkeit der Abschätzung der Schmelzenthalpie nur sehr gering ist.[1] Aktuelle Modellentwicklungen haben durch die Verwendung von Faktendatenbanken wie der Dortmunder Datenbank oder der DIPPR-Datenbank eine weitaus größere Datenbasis.
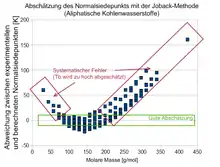
Die Formel, die für die Vorhersage des Normalsiedepunkts verwendet wird, zeigt ein anderes Problem. Joback nahm an, dass der Beitrag von Gruppen in einer homologen Reihe wie der der Alkane konstant bleibt. Dies ist jedoch keine korrekte Annahme[5]. Statt der konstanten Beiträge muss eine Abnahme der Beiträge mit zunehmender Anzahl Gruppen verwendet werden. Die Formel, die Joback auswählte, führt zu hohen Abweichungen bei kleinen und großen Molekülen und liefert eine akzeptable Abschätzung nur für mittelgroße Komponenten.
Formeln
In den folgenden Formeln bezeichnet einen Gruppenbeitrag. werden für jedes einzelne Vorkommen einer Gruppe addiert. Wenn also eine Gruppe bspw. dreimal vorkommt, wird ihr Beitrag dreimal addiert.

| Größe | Formel | Bemerkung |
|---|---|---|
| Normalsiedepunkt | ||
| Schmelzpunkt | ||
| Kritische Temperatur | Diese Gleichung benötigt einen Normalsiedepunkt Tb. Wenn ein experimenteller Wert verfügbar ist, wird empfohlen, diesen zu verwenden. Es ist andererseits auch möglich, einen mit der Joback-Methode abgeschätzten Wert zu verwenden. Dies führt jedoch zu einem größeren Fehler. | |
| Kritischer Druck | : Anzahl der Atome in der Molekülstruktur (einschließlich der Wasserstoffe) | |
| Kritisches Volumen | ||
| Bildungsenthalpie | Ideales Gas, 298 K | |
| Gibbssche Bildungsenergie | Ideales Gas, 298 K | |
| Wärmekapazität | Ideales Gas; Die Joback-Methode verwendet ein kubisches Polynom mit vier Parametern zur Beschreibung der Temperaturabhängigkeit der Wärmekapazität des idealen Gases. Die Parameter sind gültig im Bereich von 273 K bis 1000 K. | |
| Verdampfungsenthalpie am Normalsiedepunkt | ||
| Schmelzenthalpie | ||
| Dynamische Viskosität der Flüssigkeit | : Molare Masse; Die Joback-Methode verwendet zwei Parameter zur Beschreibung der Temperaturabhängigkeit der Flüssigviskosität. Die Autoren geben an, dass die Parameter bis zu einer reduzierten Temperatur gültig sind. |














Gruppenbeiträge
| Gruppe | Tc | Pc | Vc | Tb | Tm | Hf | Gf | a | b | c | d | Hm | Hv | a | b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kritischer Punkt | Phasenübergangs- temperaturen |
Kalorische Größen |
Wärmekapazität Ideales Gas |
Phasenübergangs- enthalpien |
Dynamische Viskosität | ||||||||||
| Nicht-Ring-Gruppen | |||||||||||||||
| –CH3 | 0,0141 | −0,0012 | 65 | 23,58 | −5,10 | −76,45 | −43,96 | 1,95E+1 | −8,08E−3 | 1,53E−4 | −9,67E−8 | 0,908 | 2,373 | 548,29 | −1,719 |
| –CH2– | 0,0189 | 0,0000 | 56 | 22,88 | 11,27 | −20,64 | 8,42 | −9,09E−1 | 9,50E−2 | −5,44E−5 | 1,19E−8 | 2,590 | 2,226 | 94,16 | −0,199 |
| >CH– | 0,0164 | 0,0020 | 41 | 21,74 | 12,64 | 29,89 | 58,36 | −2,30E+1 | 2,04E−1 | −2,65E−4 | 1,20E−7 | 0,749 | 1,691 | −322,15 | 1,187 |
| >C< | 0,0067 | 0,0043 | 27 | 18,25 | 46,43 | 82,23 | 116,02 | −6,62E+1 | 4,27E−1 | −6,41E−4 | 3,01E−7 | −1,460 | 0,636 | −573,56 | 2,307 |
| =CH2 | 0,0113 | −0,0028 | 56 | 18,18 | −4,32 | −9,630 | 3,77 | 2,36E+1 | −3,81E−2 | 1,72E−4 | −1,03E−7 | −0,473 | 1,724 | 495,01 | −1,539 |
| =CH– | 0,0129 | −0,0006 | 46 | 24,96 | 8,73 | 37,97 | 48,53 | −8,00 | 1,05E−1 | −9,63E−5 | 3,56E−8 | 2,691 | 2,205 | 82,28 | −0,242 |
| =C< | 0,0117 | 0,0011 | 38 | 24,14 | 11,14 | 83,99 | 92,36 | −2,81E+1 | 2,08E−1 | −3,06E−4 | 1,46E−7 | 3,063 | 2,138 | n. v. | n. v. |
| =C= | 0,0026 | 0,0028 | 36 | 26,15 | 17,78 | 142,14 | 136,70 | 2,74E+1 | −5,57E−2 | 1,01E−4 | −5,02E−8 | 4,720 | 2,661 | n. v. | n. v. |
| ≡CH | 0,0027 | −0,0008 | 46 | 9,20 | −11,18 | 79,30 | 77,71 | 2,45E+1 | −2,71E−2 | 1,11E−4 | −6,78E−8 | 2,322 | 1,155 | n. v. | n. v. |
| ≡C– | 0,0020 | 0,0016 | 37 | 27,38 | 64,32 | 115,51 | 109,82 | 7,87 | 2,01E−2 | −8,33E−6 | 1,39E−9 | 4,151 | 3,302 | n. v. | n. v. |
| Ringgruppen | |||||||||||||||
| –CH2– | 0,0100 | 0,0025 | 48 | 27,15 | 7,75 | −26,80 | −3,68 | −6,03 | 8,54E−2 | −8,00E−6 | −1,80E−8 | 0,490 | 2,398 | 307,53 | −0,798 |
| >CH– | 0,0122 | 0,0004 | 38 | 21,78 | 19,88 | 8,67 | 40,99 | −2,05E+1 | 1,62E−1 | −1,60E−4 | 6,24E−8 | 3,243 | 1,942 | −394,29 | 1,251 |
| >C< | 0,0042 | 0,0061 | 27 | 21,32 | 60,15 | 79,72 | 87,88 | −9,09E+1 | 5,57E−1 | −9,00E−4 | 4,69E−7 | −1,373 | 0,644 | n. v. | n. v. |
| =CH– | 0,0082 | 0,0011 | 41 | 26,73 | 8,13 | 2,09 | 11,30 | −2,14 | 5,74E−2 | −1,64E−6 | −1,59E−8 | 1,101 | 2,544 | 259,65 | −0,702 |
| =C< | 0,0143 | 0,0008 | 32 | 31,01 | 37,02 | 46,43 | 54,05 | −8,25 | 1,01E−1 | −1,42E−4 | 6,78E−8 | 2,394 | 3,059 | −245,74 | 0,912 |
| Halogengruppen | |||||||||||||||
| –F | 0,0111 | −0,0057 | 27 | −0,03 | −15,78 | −251,92 | −247,19 | 2,65E+1 | −9,13E−2 | 1,91E−4 | −1,03E−7 | 1,398 | −0,670 | n. v. | n. v. |
| –Cl | 0,0105 | −0,0049 | 58 | 38,13 | 13,55 | −71,55 | −64,31 | 3,33E+1 | −9,63E−2 | 1,87E−4 | −9,96E−8 | 2,515 | 4,532 | 625,45 | −1,814 |
| –Br | 0,0133 | 0,0057 | 71 | 66,86 | 43,43 | −29,48 | −38,06 | 2,86E+1 | −6,49E−2 | 1,36E−4 | −7,45E−8 | 3,603 | 6,582 | 738,91 | −2,038 |
| –I | 0,0068 | −0,0034 | 97 | 93,84 | 41,69 | 21,06 | 5,74 | 3,21E+1 | −6,41E−2 | 1,26E−4 | −6,87E−8 | 2,724 | 9,520 | 809,55 | −2,224 |
| Sauerstoffgruppen | |||||||||||||||
| –OH (Alkohole) | 0,0741 | 0,0112 | 28 | 92,88 | 44,45 | −208,04 | −189,20 | 2,57E+1 | −6,91E−2 | 1,77E−4 | −9,88E−8 | 2,406 | 16,826 | 2173,72 | −5,057 |
| –OH (Phenol) | 0,0240 | 0,0184 | −25 | 76,34 | 82,83 | −221,65 | −197,37 | −2,81 | 1,11E−1 | −1,16E−4 | 4,94E−8 | 4,490 | 12,499 | 3018,17 | −7,314 |
| –O– (Nichtring) | 0,0168 | 0,0015 | 18 | 22,42 | 22,23 | −132,22 | −105,00 | 2,55E+1 | −6,32E−2 | 1,11E−4 | −5,48E−8 | 1,188 | 2,410 | 122,09 | −0,386 |
| –O– (Ring) | 0,0098 | 0,0048 | 13 | 31,22 | 23,05 | −138,16 | −98,22 | 1,22E+1 | −1,26E−2 | 6,03E−5 | −3,86E−8 | 5,879 | 4,682 | 440,24 | −0,953 |
| >C=O (Nichtring) | 0,0380 | 0,0031 | 62 | 76,75 | 61,20 | −133,22 | −120,50 | 6,45 | 6,70E−2 | −3,57E−5 | 2,86E−9 | 4,189 | 8,972 | 340,35 | −0,350 |
| >C=O (Ring) | 0,0284 | 0,0028 | 55 | 94,97 | 75,97 | −164,50 | −126,27 | 3,04E+1 | −8,29E−2 | 2,36E−4 | −1,31E−7 | n. v. | 6,645 | n. v. | n. v. |
| O=CH– (Aldehyd) | 0,0379 | 0,0030 | 82 | 72,24 | 36,90 | −162,03 | −143,48 | 3,09E+1 | −3,36E−2 | 1,60E−4 | −9,88E−8 | 3,197 | 9,093 | 740,92 | −1,713 |
| –COOH (Säure) | 0,0791 | 0,0077 | 89 | 169,09 | 155,50 | −426,72 | −387,87 | 2,41E+1 | 4,27E−2 | 8,04E−5 | −6,87E−8 | 11,051 | 19,537 | 1317,23 | −2,578 |
| –COO– (Ester) | 0,0481 | 0,0005 | 82 | 81,10 | 53,60 | −337,92 | −301,95 | 2,45E+1 | 4,02E−2 | 4,02E−5 | −4,52E−8 | 6,959 | 9,633 | 483,88 | −0,966 |
| =O (andere als oben) | 0,0143 | 0,0101 | 36 | −10,50 | 2,08 | −247,61 | −250,83 | 6,82 | 1,96E−2 | 1,27E−5 | −1,78E−8 | 3,624 | 5,909 | 675,24 | −1,340 |
| Stickstoffgruppen | |||||||||||||||
| –NH2 | 0,0243 | 0,0109 | 38 | 73,23 | 66,89 | −22,02 | 14,07 | 2,69E+1 | −4,12E−2 | 1,64E−4 | −9,76E−8 | 3,515 | 10,788 | n. v. | n. v. |
| >NH (Nichtring) | 0,0295 | 0,0077 | 35 | 50,17 | 52,66 | 53,47 | 89,39 | −1,21 | 7,62E−2 | −4,86E−5 | 1,05E−8 | 5,099 | 6,436 | n. v. | n. v. |
| >NH (Ring) | 0,0130 | 0,0114 | 29 | 52,82 | 101,51 | 31,65 | 75,61 | 1,18E+1 | −2,30E−2 | 1,07E−4 | −6,28E−8 | 7,490 | 6,930 | n. v. | n. v. |
| >N–(Nichtring) | 0,0169 | 0,0074 | 9 | 11,74 | 48,84 | 123,34 | 163,16 | −3,11E+1 | 2,27E−1 | −3,20E−4 | 1,46E−7 | 4,703 | 1,896 | n. v. | n. v. |
| –N= (Nichtrring) | 0,0255 | −0,0099 | n. v. | 74,60 | n. v. | 23,61 | n. v. | n. v. | n. v. | n. v. | n. v. | n. v. | 3,335 | n. v. | n. v. |
| –N= (Ring) | 0,0085 | 0,0076 | 34 | 57,55 | 68,40 | 93,70 | 119,66 | 5,69 | −4,12E−3 | 1,28E−4 | −8,88E−8 | 3,649 | 6,528 | n. v. | n. v. |
| =NH | n. v. | n. v. | n. v. | 83,08 | 68,91 | 93,70 | 119,66 | 5,69 | −4,12E−3 | 1,28E−4 | −8,88-8 | n. v. | 12,169 | n. v. | n. v. |
| –CN | 0,0496 | −0,0101 | 91 | 125,66 | 59,89 | 88,43 | 89,22 | 3,65E+1 | −7,33E−2 | 1,84E−4 | −1,03E−7 | 2,414 | 12,851 | n. v. | n. v. |
| –NO2 | 0,0437 | 0,0064 | 91 | 152,54 | 127,24 | −66,57 | −16,83 | 2,59E+1 | −3,74E−3 | 1,29E−4 | −8,88E−8 | 9,679 | 16,738 | n. v. | n. v. |
| Schwefelgruppen | |||||||||||||||
| –SH | 0,0031 | 0,0084 | 63 | 63,56 | 20,09 | −17,33 | −22,99 | 3,53E+1 | −7,58E−2 | 1,85E−4 | −1,03E−7 | 2,360 | 6,884 | n. v. | n. v. |
| –S– (Nichtring) | 0,0119 | 0,0049 | 54 | 68,78 | 34,40 | 41,87 | 33,12 | 1,96E+1 | −5,61E−3 | 4,02E−5 | −2,76E−8 | 4,130 | 6,817 | n. v. | n. v. |
| –S– (Ring) | 0,0019 | 0,0051 | 38 | 52,10 | 79,93 | 39,10 | 27,76 | 1,67E+1 | 4,81E−3 | 2,77E−5 | −2,11E−8 | 1,557 | 5,984 | n. v. | n. v. |
Beispielrechnung
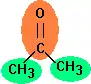
Aceton (Propanon) ist das einfachste Keton und wird nach der Joback-Methode in drei Gruppen aufgeteilt: Zwei Methylgruppen (–CH3) und eine Ketogruppe (C=O). Da die Methylgruppe zweimal vorkommt, wird ihr Beitrag doppelt addiert.
| –CH3 | >C=O (Nichtring) | ||||||
|---|---|---|---|---|---|---|---|
| Eigenschaft | Anzahl der Gruppen |
Beitrag | Anzahl der Gruppen |
Beitrag | Berechneter Wert | Einheit | |
| Tc | 2 | 0,0141 | 1 | 0,0380 | 0,0662 | 500,5590 | K |
| Pc | 2 | −1,20E−03 | 1 | 3,10E−03 | 7,00E−04 | 48,0250 | bar |
| Vc | 2 | 65,0000 | 1 | 62,0000 | 192,0000 | 209,5000 | cm3/mol |
| Tb | 2 | 23,5800 | 1 | 76,7500 | 123,9100 | 322,1100 | K |
| Tm | 2 | −5,1000 | 1 | 61,2000 | 51,0000 | 173,5000 | K |
| Hf | 2 | −76,4500 | 1 | −133,2200 | −286,1200 | −217,8300 | kJ/mol |
| Gf | 2 | −43,9600 | 1 | −120,5000 | −208,4200 | −154,5400 | kJ/mol |
| Cpa | 2 | 1,95E+01 | 1 | 6,45E+00 | 4,55E+01 | ||
| Cpb | 2 | −8,08E−03 | 1 | 6,70E−02 | 5,08E−02 | ||
| Cpc | 2 | 1,53E−04 | 1 | −3,57E−05 | 2,70E−04 | ||
| Cpd | 2 | −9,67E−08 | 1 | 2,86E−09 | −1,91E−07 | ||
| Cp | bei T = 300 K | 75,3264 | J/(mol·K) | ||||
| Hm | 2 | 0,9080 | 1 | 4,1890 | 6,0050 | 5,1250 | kJ/mol |
| Hv | 2 | 2,3730 | 1 | 8,9720 | 13,7180 | 29,018 | kJ/mol |
| ηa | 2 | 548,2900 | 1 | 340,3500 | 1436,9300 | ||
| ηb | 2 | −1,7190 | 1 | −0,3500 | −3,7880 | ||
| η | bei T = 300 K | 0,0002942 | Pa s | ||||

Weblinks
Einzelnachweise
- K. G Joback, R. C Reid: Estimation of pure-component properties from group-contributions. In: Chemical Engineering Communications. Band 57, Nr. 1, 1987, S. 233–243.
- A. L Lydersen, R. A Greenkorn, O. A Hougen: Estimation of Critical Properties of Organic Compounds by the Method of Group Contributions. In: Engineering Experiment Station Report. University of Wisconsin, Madison, Wisconsin 1955.
- Leonidas Constantinou, Rafiqul Gani: New group contribution method for estimating properties of pure compounds. In: AIChE Journal. Band 40, Nr. 10, Oktober 1994, S. 1697–1710, doi:10.1002/aic.690401011.
- Yash Nannoolal, Jürgen Rarey, Deresh Ramjugernath: Estimation of pure component properties: Part 2. Estimation of critical property data by group contribution. In: Fluid Phase Equilibria. Band 252, Nr. 1–2, 1. Februar 2007, S. 1–27, doi:10.1016/j.fluid.2006.11.014.
- S. E. Stein, R. L. Brown: Estimation of normal boiling points from group contributions. In: Journal of Chemical Information and Computer Sciences. Band 34, Nr. 3, 1. April 1994, S. 581–587, doi:10.1021/ci00019a016.