Diamond-Blackfan-Syndrom
Das Diamond-Blackfan-Syndrom (lat. Erythrogenesis imperfecta), auch Diamond-Blackfan-Anämie (DBA) oder chronische kongenitale hypoplastische Anämie genannt, ist eine schwere chronische Blutarmut mit einer zu geringen Anzahl an roten Blutkörperchen, die bei Betroffenen meist bereits während des ersten Lebensjahrs auftritt.
Ihre Ursache hat die anämische Erkrankung, eine Sonderform einer aplastischen Anämie, in der selektiven Störung der Bildung der Erythrozyten (roten Blutkörperchen) im Knochenmark.[1] Bei etwa 25 % der Betroffenen treten zudem andere Erbkrankheiten bzw. Fehlbildungen auf (siehe Diagnose).
| Klassifikation nach ICD-10 | |
|---|---|
| D61.0 | Angeborene aplastische Anämie - Blackfan-Diamond-Anämie |
| ICD-10 online (WHO-Version 2019) | |
Geschichte
Louis Diamond und Kenneth Blackfan beschrieben die erbliche hypoplastische Anämie erstmals 1938.[2] 1961 präsentierten Diamond und seine Mitarbeiter eine Längsschnittstudie über 25 Jahre, die auf den Daten von 30 Patienten basiert und stellten dabei einen Zusammenhang mit Abnormalitäten im Skelettaufbau fest.[3]
1997 wurde auf dem Chromosom 19 der Bereich identifiziert, der das entsprechende mutierte Gen des Diamond-Blackfan-Syndroms trägt.[4][5]
1999 wurde festgestellt, dass Mutationen in dem ribosomalen Protein-S19-Gen (RPS19) für die Erkrankung von 42 von 172 DBS-Patienten verantwortlich sind.[6]
2001 wurde entdeckt, dass ein zweites für das Diamond-Blackfan-Syndrom verantwortliches Gen auf dem Chromosom 8 liegt.[7]
Ätiologie
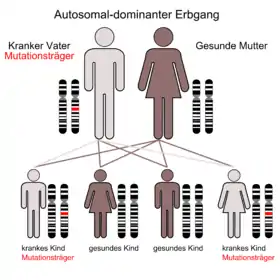
Die Krankheit kann autosomal-rezessiv oder -dominant erblich sein. Die Veränderung befindet sich dabei auf dem langen Arm des Chromosoms 19 an der Position 13. Bei den meisten Betroffenen tritt die Erkrankung sporadisch auf. Bei etwa 15 % der Betroffenen wurde die Erkrankung von den Eltern an ihre Kinder vererbt.[1] Die eigentliche Ursache für das Auftreten der Erkrankung ist bisher nicht bekannt. Es wird vermutet, dass eine angeborene Anomalie der erythrozytären Stammzellen für die Erkrankung verantwortlich sein könnte. Durch diese Anomalie lassen sich die erythrozytären Stammzellen im Knochenmark nicht mehr genügend durch Erythropoetin zur Zellteilung anregen, sodass bei Betroffenen eine Blutarmut mit einer zu geringen Zahl an roten Blutkörperchen auftritt.
Auftreten
Das Diamond-Blackfan-Syndrom ist eine ausgesprochen seltene Krankheit. Mit einer Häufigkeit von 0,3 Erkrankungen auf 100.000 Personen sind aplastische Anämien (inklusive der Sonderformen Fanconi-Anämie und Diamond-Blackfan-Syndrom) in Europa so selten, dass die meisten Mediziner mit dieser Erkrankung während ihrer gesamten Arbeitszeit nie konfrontiert werden. Die Häufigkeit der DBA liegt bei ca. 5 – 7 Fällen pro 1 Million Lebendgeburten.[1]
Weltweit sind etwa 800 Fälle beschrieben, in Deutschland gibt es zurzeit ca. 130 Patienten.[8]
Symptome
Die Blutarmut zeigt sich meist in den ersten sechs Lebensmonaten, bei etwa der Hälfte der Fälle bereits vor dem dritten Lebensmonat, bei etwa einem Drittel der Fälle sogar bereits bei der Geburt oder im ersten Lebensmonat mit Blässe. Häufig können bei Betroffenen eines Diamond-Blackfan-Syndroms Fehlbildungen des Körpers vorhanden sein, wie beispielsweise eine Lippen-Kiefer-Gaumenspalte, ein kleiner Schädel (Mikrocephalus), kleine Augäpfel (Mikrophthalmus), weit auseinanderstehende Augen (Hypertelorismus) und ein hoher Gaumen. Die Hälfte der Betroffenen eines Diamond-Blackfan-Syndroms sind kleinwüchsig. Ein Drittel der Betroffenen hat einen Herzfehler, Missbildungen der Nieren oder Fehlbildungen der Finger, vor allem des Daumens. Zudem kann die geistige Entwicklung betroffener Kinder verzögert sein.
Die Kombination aus angeborener hypoplastischer Anämie und bestimmten Fehlbildungen – wie Dreigliedrigkeit (Triphalangie) des Daumens (Triphalangealer Daumen), Hydrozephalus, Lippen-Kiefer-Gaumenspalte und multiplen Gelenkkontrakturen – wird Aase-Syndrom oder Aase-Smith-Syndrom II genannt. Ob es sich tatsächlich um ein eigenständiges Krankheitsbild oder eine Variante des Diamond-Blackfan-Syndroms handelt, ist unklar.[9]
Diagnose
Charakteristisch ist ein Erythroblastenmangel („kongenitale Erythroblastophthise“; Erythroblastophthise = Schwund des erythropoetischen Gewebes im Knochenmark, eine aplastische Anämie im engeren Sinne[10]) mit Retikulozytopenie, der eventuell mit Fehlbildungen des Geschlechtstraktes, pseudomongoloidem Habitus (typischer Gesichtsausdruck), Mikrozephalus (abnorme Kleinheit des Hirnschädels), Mikrophthalmus („kleines Auge“), Hypertelorismus (großer Augenabstand), hohem Gaumen (Gotischer Gaumen), sowie einem Rückstand der geistigen und körperlichen Entwicklung begleitet wird.[11]
Während bei Gesunden der Hämoglobin-Wert (Hb) im Blut bei über 11 g/dl liegt, kann er bei einer DBA unter 6 g/dl sein. Die Diagnose DBA kann nach der Analyse des Blutes mit typischen Veränderungen durch eine Knochenmarkpunktion bestätigt werden. Bei DBA-Patienten findet man im Knochenmark keine oder nur wenig heranreifende Vorläuferzellen der roten Blutkörperchen.[1]
Bei etwa 20 bis 25 % der DBA-Patienten kann die Krankheit per Gentest über die Mutation des RPS19-Gens diagnostiziert werden.
Therapie
Die Krankheit kann mit Corticosteroiden gut therapiert werden. In einer Studie sprachen 82 % der Patienten mit der Bildung von roten Blutkörperchen auf die Corticosteroid-Therapie anfänglich an.[12] Diese Corticosteroid-Behandlung sollte aber erst nach dem ersten Lebensjahr begonnen werden.
Im ersten Lebensjahr oder bei Patienten, die nicht auf die Corticosteroid-Behandlung ansprechen, werden Bluttransfusionen zur Behandlung der DBA eingesetzt. Dabei ist eine Anhäufung von Eisen im Körper, eine sogenannte Hämosiderose, ab dem dritten Lebensjahr mit sogenannten Chelatbildnern wie Deferoxamin vorzubeugen. Es ist möglich, dass bei der Behandlung mit Transfusionen und Corticosteroiden Phasen der Remission auftreten, während derer keine Transfusionen oder Steroidgaben notwendig sind.
Eine Stammzelltransplantation (Knochenmarktransplantation (KMT)) kann die Blutarmut der DBA heilen. Diese Maßnahme wird insbesondere bei Patienten in Betracht gezogen, die abhängig von Transfusionen sind, da häufige Transfusionen zu Organschäden führen können. Die KMT ist zurzeit die einzige Therapieform, mit der man DBA heilen kann. Das Finden eines passenden Knochenmarkspenders ist schwierig und das Risiko einer Knochenmarktransplantation von einem Nicht-Familienspender ist so hoch, dass über eine solche Therapie im Einzelfall entschieden werden muss.[1] Eine Heilung durch Behebung der Ursache der Erkrankung ist nicht möglich, da die Ursache bisher nicht bekannt ist und Veränderungen der Chromosomen bisher nicht rückgängig gemacht werden können. Nur in wenigen Fällen wurden bisher Spontanheilungen des Diamond-Blackfan-Syndroms beschrieben.
Einzelnachweise
- Uniklinik Freiburg: Diamond-Blackfan-Anämie – eine seltene angeborene Knochenmarkerkrankung, Internet Archive abgerufen am 7. August 2013
- L. K. Diamond, K. D. Blackfan: Hypoplastic anemia. In: Am. J. Dis. Child. 56/1938, S. 464–467.
- L. K. Diamond, D. W. Allen, F. B. Magill: Congenital (erythroid) hypoplastic anemia: a 25 year study. In: Am. J. Dis. Child. 102/1961, S. 403–415.
- P. Gustavsson u. a.: Diamond-Blackfan anaemia: genetic homogeneity for a gene on chromosome 19q13 restricted to 1.8 Mb. In: Nat. Genet. 16/1997, S. 368–371.
- P. Gustavsson u. a.: Diamond-Blackfan anaemia in a girl with a de novo balanced reciprocal X;19 translocation. In: J. Med. Genet. 34/1997, S. 779–782.
- N. Draptchinskaia u. a.: The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. In: Nat. Genet. 21/1999, S. 168–175.
- H. Gazda u. a.: Evidence for linkage of familial Diamond-Blackfan anemia to chromosome 8p23.3-p22 and for non-19q non-8p disease. In: Blood. 97/2001, S. 2145–2150.
- www.diamond-blackfan.de abgerufen am 7. August 2007
- Aase-Smith syndrome bei whonamedit.com
- Ludwig Heilmeyer, Herbert Begemann: Blut und Blutkrankheiten. In: Ludwig Heilmeyer (Hrsg.): Lehrbuch der Inneren Medizin. Springer-Verlag, Berlin/Göttingen/Heidelberg 1955; 2. Auflage ebenda 1961, S. 376–449, hier: S. 416: Die aplastische Anämie im engeren Sinne (Erythrobladtophthise).
- Ärztliche Praxis: D.-Blackfan-Syndrom (Memento vom 28. September 2007 im Internet Archive)
- A. Vlachos u. a.: The Diamond Blackfan Anemia Registry: tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. In: J. Pediatr. Hematol. Oncol. 23/2001, S. 377–382.
Literatur
- Friedrich Carl Sitzmann: Pädiatrie. (= Duale Reihe). 2. Auflage. Georg Thieme Verlag, Stuttgart 2002, ISBN 3-13-125332-0, S. 455.
Weblinks
- Diamond-Blackfan-Syndrom. In: Orphanet (Datenbank für seltene Krankheiten).
- DBA-Selbsthilfegruppe e.V.
- Diamond-Blackfan and you (in Englisch)
- Netzwerk für angeborene Störungen der Blutbildung