Chromosome conformation capture
Chromosome conformation capture (dt. "Konformationserfassung von Chromosomen", oft abgekürzt mit 3C-Technologien oder 3C-basierte Methoden[1]) sind eine Reihe von molekularbiologischen Methoden, mit denen die räumliche Organisation von Chromatin in einer Zelle analysiert wird. Diese Methoden quantifizieren die Anzahl der Wechselwirkungen zwischen genomischen Loci, die sich im dreidimensionalen Raum in unmittelbarer Nähe befinden, aber im linearen Genom durch viele Nukleotide getrennt sein können.[2] Solche Wechselwirkungen können sich aus biologischen Funktionen, wie z. B. Promotor-Enhancer-Wechselwirkungen, oder aus einer zufälligen Schleifenbildung ergeben, bei der eine ungerichtete physikalische Bewegung des Chromatins zu einer Kollision der Loci führt.[3] Die Frequenz dieser Interaktionen können direkt analysiert werden,[4] oder sie können in Abstände umgewandelt und zur Rekonstruktion von 3D-Strukturen verwendet werden.[5] Diese Technologie hat die genetische und epigenetische Untersuchung von Chromosomen sowohl in Modellorganismen als auch beim Menschen weiter unterstützt.
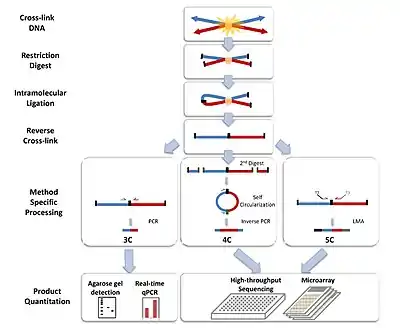
Der Hauptunterschied zwischen 3C-basierten Methoden besteht in ihrem Umfang. Wenn beispielsweise PCR zum Nachweis von Wechselwirkungen in einem 3C-Experiment verwendet wird, dann werden die Wechselwirkungen zwischen zwei spezifischen Fragmenten quantifiziert. Im Gegensatz dazu quantifiziert die Hi-C-Methode die Wechselwirkungen zwischen allen möglichen Paaren von Fragmenten gleichzeitig.
Experimentelle Methoden
Alle 3C-Methoden beginnen mit einer ähnlichen Menge von Schritten, die an einer Zellprobe durchgeführt werden.
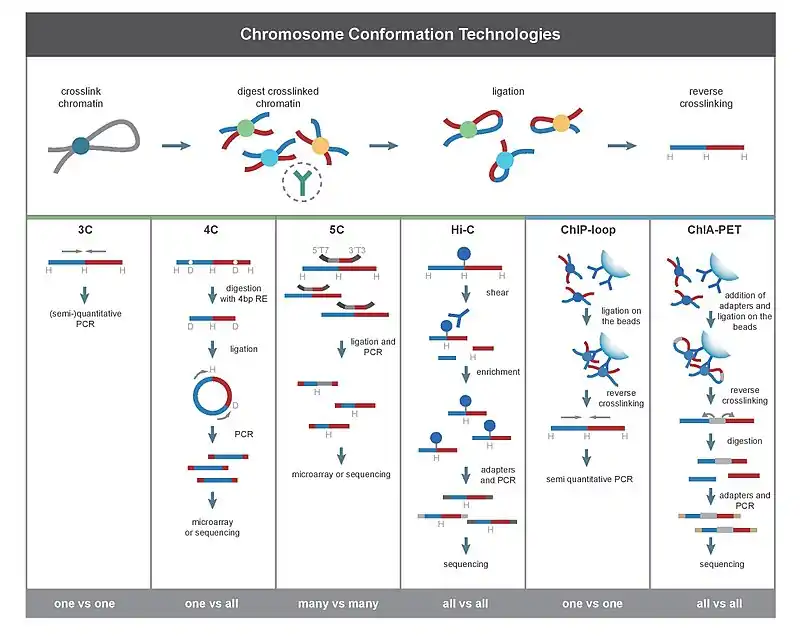
Zunächst werden die Zellgenome mit Formaldehyd[6] vernetzt. Dies führt Bindungen ein, die die Wechselwirkungen zwischen genomischen Orten "einfrieren". Die Behandlung von Zellen mit 1-3 % Formaldehyd für 10-30 min bei Raumtemperatur ist am weitesten verbreitet. Jedoch ist eine Standardisierung zur Verhinderung einer proteinreichen DNA-Vernetzung notwendig, da dies die Effizienz des Restriktionsverdaus im nachfolgenden Schritt negativ beeinflussen kann. Das Genom wird dann mit einer Restriktionsendonuklease in Fragmente zerlegt. Dazu werden Restriktionsenzyme verwendet, die Erkennungssequenzen mit einer Länge von 6bp wie EcoR1 oder HindIII schneiden. Diese schneiden das Genom ca. alle 4000bp. Damit ergeben sich etwa 1 Million Fragmente im menschlichen Genom.[7][8] Die Größe der Restriktionsfragmente bestimmt die Auflösung des Interaktionsmappings. Für eine präzisere Abbildung der Interaktionen kann auch ein Restriktionsenzym von 4bp verwendet verwendet werden.
Als nächstes erfolgt ein Schritt zur zufälligen Verknüpfung (Ligation) der Fragmente. Die Endstücke der Fragmente können durch komplementäre Basenpaarung dazu gebracht werden, sich zu verbinden. Dies geschieht bei niedrigen DNA-Konzentrationen in Gegenwart von T4-DNA-Ligase[9]. Hierbei wird die Verknüpfung zwischen vernetzten, interagierenden Fragmenten gegenüber der Verknüpfung zwischen nicht vernetzten Fragmenten bevorzugt. Anschließend werden interagierende Loci durch Amplifikation ligierter Verbindungen mit PCR-Methoden quantifiziert.[10][11]
3C (one-vs-one)
Das Experiment zur Chromosome Confirmation Capture (3C) quantifiziert die Wechselwirkungen zwischen einem einzigen Paar genomischer Loci. So kann beispielsweise mit 3C die mögliche Interaktion zwischen Promotor und Enhancer getestet werden. Ligierte Fragmente werden mittels PCR mit bekannten Primern nachgewiesen.
4C (one-vs-all)
Chromosome conformation capture-on-chip (4C) erfasst Interaktionen zwischen einem Locus und allen anderen genomischen Loci. Es handelt sich um einen zweiten Ligationsschritt, um selbstzirkuläre DNA-Fragmente zu erzeugen, die zur Durchführung der inversen PCR verwendet werden. Die inverse PCR ermöglicht es, eine bekannte Sequenz zur Amplifikation der daran gebundenen unbekannten Sequenz zu verwenden.[2][12] Im Gegensatz zu den 3C- und 5C-Methoden erfordert die 4C-Technik keine Vorkenntnisse über beide interagierenden chromosomalen Regionen. Die mit 4C erhaltenen Ergebnisse sind bei den meisten der Wechselwirkungen, die zwischen den nahegelegenen Bereichen erkannt werden, hochgradig reproduzierbar. Auf einem einzelnen Microarray können etwa eine Million Interaktionen analysiert werden.
5C (many-vs-many)
Chromosome conformation capture carbon copy (5C) erkennt Wechselwirkungen zwischen allen Restriktionsfragmenten innerhalb eines bestimmten Bereichs, wobei die Größe dieses Bereichs typischerweise nicht größer als eine Megabase ist. 5C hat jedoch eine relativ geringe Abdeckung. Die 5C-Technik überwindet einige Probleme im intramolekularen Ligaturschritt und ist nützlich für die Konstruktion komplexer Wechselwirkungen der untersuchten Loci. Dieser Ansatz ist ungeeignet für die Durchführung genomweiter komplexer Interaktionen, da dafür Millionen von 5C-Primern verwendet werden müssten.
Hi-C (all-vs-all)
Hi-C verwendet eine Hochdurchsatz-Sequenzierung, um die Nukleotidsequenz von Fragmenten zu finden.[2][13] Das ursprüngliche Protokoll verwendete die Schrotschuss-Sequenzierung, die eine kurze Sequenz von jedem Ende jedes ligierten Fragments abruft. Daher sollten die beiden erhaltenen Sequenzen für ein bestimmtes ligiertes Fragment zwei verschiedene Restriktionsfragmente repräsentieren, die im Zufallsligaturschritt zusammen ligiert wurden. Das Paar von Sequenzen ist individuell auf das Genom ausgerichtet und bestimmt so die an diesem Ligaturereignis beteiligten Fragmente. Daher werden alle möglichen paarweisen Wechselwirkungen zwischen den Fragmenten getestet.
Forscher versuchen, das Möglichkeiten der Hi-C-Detektion durch eine Studie zu untersuchen, die sich auf das Screening primärer Hirntumore konzentriert.[14] Vor solchen Tumoruntersuchungen konzentrierte sich Hi-C vor allem auf die Analyse von Zelllinien.[15]
Methoden basierend auf Sequenzerfassung
Eine Reihe von Methoden verwenden die Erfassung von Oligonukleotiden, um 3C- und Hi-C-Bibliotheken für bestimmte Loci von Interesse anzureichern.[16] Zu diesen Methoden gehören Capture-C,[17] NG Capture-C,[18] Capture-3C[19] und Capture Hi-C.[20] Diese Methoden sind in der Lage, eine höhere Auflösung und Empfindlichkeit zu erzeugen als 4C-basierte Methoden.
Methoden für einzelne Zellen
Mit Single-Cell Hi-C können auch die auftretenden Wechselwirkungen in einzelnen Zellen untersucht werden.[21][22]
ChIP-loop
ChIP-loop kombiniert 3C mit ChIP-seq, um Wechselwirkungen zwischen zwei zu untersuchenden Loci zu erkennen, die durch ein Protein vermittelt werden.[2][23] ChIP-loop kann nützlich sein, um weiträumige cis-Interaktionen und trans-Interaktionen zu identifizieren, die durch Proteine vermittelt werden, da häufige DNA-Kollisionen nicht auftreten.
Genomweite Methoden
ChIA-PET kombiniert Hi-C mit ChIP-Seq, um alle Interaktionen zu erkennen, die durch ein zu untersuchendes Protein vermittelt werden. HiChIP wurde entwickelt, um eine ähnliche Analyse wie ChIA-PET mit weniger Ausgangsmaterial zu ermöglichen.
Biologische Auswirkungen
3C-Methoden haben zu einer Reihe biologischer Erkenntnisse geführt, darunter die Entdeckung neuer struktureller Merkmale von Chromosomen, die Katalogisierung von Chromatinschleifen und ein besseres Verständnis der transkriptionellen Regulationsmechanismen (deren Störung zu Krankheiten führen kann).[24]
3C-Methoden haben die Bedeutung der räumlichen Nähe von regulatorischen Elementen zu den Genen, die sie regulieren, gezeigt. So bildet beispielsweise in Geweben, die Globin-Gene exprimieren, die Kontrollregion β-Globin-Locus eine Schleife mit diesen Genen. Diese Schleife findet sich nicht in Geweben, in denen das Gen nicht exprimiert wird.[25] Diese Technologie hat die genetische und epigenetische Untersuchung von Chromosomen sowohl in Modellorganismen als auch beim Menschen weiter unterstützt.
Diese Methoden haben eine groß angelegte Organisation des Genoms in Topologically Associating Domains (TADs) ergeben, die mit epigenetischen Markern korrelieren. Einige TADs sind transkriptionell aktiv, während andere unterdrückt werden.[26] Viele TADs wurden in D. melanogaster, Maus und Mensch gefunden.[27] Darüber hinaus spielen die Proteine CTCF und Cohesin eine wichtige Rolle bei der Bestimmung von TADs und Enhancer-Promoter-Interaktionen. Das Ergebnis zeigt, dass die Ausrichtung der CTCF-Bindungsmotive in einer Enhancer-Promoter-Schleife einander zugewandt sein sollte, damit der Enhancer sein richtiges Ziel findet.[28]
Menschliche Krankheiten
Es gibt mehrere Krankheiten, die durch fehlerhafte Wechselwirkungen zwischen Promotor und Enhancer verursacht werden.[29]
- Beta-Thalassämie ist eine bestimmte Art von Bluterkrankungen, die durch eine Deletion des LCR-Enhancers verursacht werden.[30][31]
- Holoprosencephalie ist eine Kopfschmerzerkrankung, die durch eine Mutation im SBE2-Enhancer-Element verursacht wird. Dies schwächt wiederum die Produktion des SHH-Gens.[32]
- Das Adenokarzinom der Lunge kann durch eine Duplikation des Enhancers für das MYC-Gen verursacht werden.[33]
- Die adulte T-Zell-Leukämie wird durch die Einführung eines neuen Enhancers verursacht.[34]
Datenanalyse
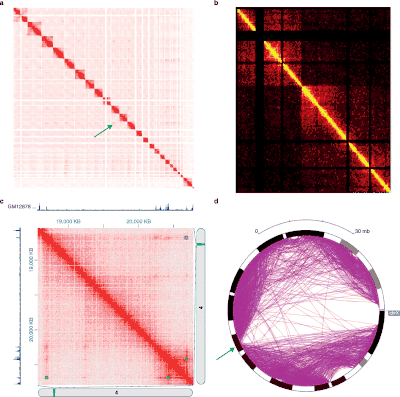
Die verschiedenen Experimente mit der 3C-Methode erzeugen Daten mit sehr unterschiedlichen Strukturen und statistischen Eigenschaften. Daher gibt es für jeden Experimenttyp spezifische Analysepakete.[40]
Hi-C-Daten werden häufig verwendet, um die genomweite Chromatinorganisation zu analysieren, wie beispielsweise topologically associating domains (TADs), linear zusammenhängende Bereiche des Genoms, die im dreidimensionalen Raum im Zellkern nahe zusammen sind.[41] Es wurden mehrere Algorithmen entwickelt, um TADs aus Hi-C-Daten zu identifizieren.[42][43]
Hi-C und seine nachfolgenden Analysen entwickeln sich weiter. Fit-Hi-C[3] ist ein Verfahren, das auf einem diskreten Binning-Ansatz mit Modifikationen des addierten Interaktionsabstandes (initiale Spline-Fitting, alias Spline-1) und der Verfeinerung des Nullmodells (Spline-2) basiert. Das Ergebnis von Fit-Hi-C ist eine Liste von paarweisen intra-chromosomalen Wechselwirkungen mit ihren p-Werten und q-Werten.[44]
Ein wesentlicher Störfaktor in 3C-Technologien sind die häufigen unspezifischen Wechselwirkungen zwischen genomischen Loci, die aufgrund von zufälligem Polymerverhalten auftreten. Eine Interaktion zwischen zwei Loci muss durch statistische Signifikanzprüfungen als spezifisch bestätigt werden.[3]
Normalisierung der Hi-C-Kontaktkarte
Es gibt zwei Hauptarten der Normalisierung von rohen Hi-C-Kontakt-Heatmaps. Der erste Weg ist die Annahme einer gleichmäßigen Sichtbarkeit, d. h. es besteht die gleiche Chance für jede chromosomale Position, eine Interaktion zu haben. Daher sollte das wahre Signal einer Hi-C-Kontaktkarte eine symmetrische Matrix sein (die symmetrische Matrix hat konstante Zeilen- und Spaltensummen). Ein Beispiel für Algorithmen, die von gleicher Sichtbarkeit ausgehen, ist der Sinkhorn-Knopp-Algorithmus, der die ursprüngliche Hi-C-Kontaktkarte in eine ausgewogene Matrix skaliert.
Die andere Variante ist anzunehmen, dass jeder chromosomalen Position ein Bias zugeordnet ist. Der Wert in der Kontaktkarte an jeder Koordinate ist das echte Signal multipliziert dem Bias, der den beiden Kontaktpositionen zugeordnet sind. Ein Beispiel für Algorithmen, die darauf abzielen, dieses Modell der Verzerrung zu lösen, ist die iterative Korrektur, die schrittweise die Zeilen- und Spaltenverzerrung aus der rohen Hi-C-Kontaktkarte entfernt. Für die Analyse von Hi-C-Daten stehen eine Reihe von Software-Tools zur Verfügung.[45]
DNA-Motivanalyse
DNA-Motive sind spezifische kurze DNA-Sequenzen, oft 8-20 Nukleotide lang,[46] die in einem Satz von Sequenzen mit einer gemeinsamen biologischen Funktion statistisch überrepräsentiert sind. Derzeit (2019) sind regulatorische Motive bei weiträumigen Chromatin-Interaktionen noch nicht umfassend untersucht worden. Mehrere Studien haben sich darauf konzentriert, die Auswirkungen von DNA-Motiven auf die Interaktion zwischen Promotor und Enhancer zu untersuchen.
Bailey et al. haben identifiziert, dass das ZNF143-Motiv in den Promotorregionen eine Spezifität für die Sequenzen für Promotor-Enhancer-Interaktionen bietet.[47] Die Mutation des ZNF143-Motivs verminderte die Häufigkeit von Promotor-Enhancer-Interaktionen, was darauf hindeutet, dass ZNF143 ein neuartiger Faktor für Chromatin-Schleifenbildung ist.
Analyse von Krebs-Genomen
Die 3C-basierten Techniken können Einblicke in die chromosomalen Umlagerungen im Krebsgenom geben.[48] Darüber hinaus können sie Veränderungen der räumlichen Nähe von regulatorischen Elementen und ihren Zielgenen zeigen, die ein tieferes Verständnis der strukturellen und funktionellen Grundlagen des Genoms ermöglichen.[49]
Einzelnachweise
- de Wit E, de Laat W: A decade of 3C technologies: insights into nuclear organization. In: Genes & Development. 26, Nr. 1, Januar 2012, S. 11–24. doi:10.1101/gad.179804.111. PMID 22215806. PMC 3258961 (freier Volltext).
- Hakim O, Misteli T: SnapShot: Chromosome confirmation capture. In: Cell. 148, Nr. 5, März 2012, S. 1068.e1–2. doi:10.1016/j.cell.2012.02.019. PMID 22385969. PMC 6374129 (freier Volltext).
- Ay F, Bailey TL, Noble WS: Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. In: Genome Research. 24, Nr. 6, Juni 2014, S. 999–1011. doi:10.1101/gr.160374.113. PMID 24501021. PMC 4032863 (freier Volltext).
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. In: Cell. 159, Nr. 7, Dezember 2014, S. 1665–80. doi:10.1016/j.cell.2014.11.021. PMID 25497547. PMC 5635824 (freier Volltext).
- Varoquaux N, Ay F, Noble WS, Vert JP: A statistical approach for inferring the 3D structure of the genome. In: Bioinformatics. 30, Nr. 12, Juni 2014, S. i26–33. doi:10.1093/bioinformatics/btu268. PMID 24931992. PMC 4229903 (freier Volltext).
- Alexey Gavrilov, Elvira Eivazova, Iryna Pirozhkova, Marc Lipinski, Sergey Razin: Chromosome Conformation Capture (from 3C to 5C) and Its ChIP-Based Modification. In: Chromatin Immunoprecipitation Assays. Band 567. Humana Press, Totowa, NJ 2009, ISBN 978-1-60327-413-5, S. 171–188, doi:10.1007/978-1-60327-414-2_12 (springer.com [abgerufen am 21. Juli 2019]).
- Naumova N, Smith EM, Zhan Y, Dekker J: Analysis of long-range chromatin interactions using Chromosome Conformation Capture. In: Methods. 58, Nr. 3, November 2012, S. 192–203. doi:10.1016/j.ymeth.2012.07.022. PMID 22903059. PMC 3874837 (freier Volltext).
- Belton JM, Dekker J: Chromosome Conformation Capture (3C) in Budding Yeast. In: Cold Spring Harbor Protocols. 2015, Nr. 6, Juni 2015, S. 580–6. doi:10.1101/pdb.prot085175. PMID 26034304.
- Gavrilov AA, Golov AK, Razin SV: Actual ligation frequencies in the chromosome conformation capture procedure. In: PLOS ONE. 8, Nr. 3, 26. März 2013, S. e60403. bibcode:2013PLoSO...860403G. doi:10.1371/journal.pone.0060403. PMID 23555968. PMC 3608588 (freier Volltext).
- Naumova N, Smith EM, Zhan Y, Dekker J: Analysis of long-range chromatin interactions using Chromosome Conformation Capture. In: Methods. 58, Nr. 3, November 2012, S. 192–203. doi:10.1016/j.ymeth.2012.07.022. PMID 22903059. PMC 3874837 (freier Volltext).
- Gavrilov AA, Golov AK, Razin SV: Actual ligation frequencies in the chromosome conformation capture procedure. In: PLOS ONE. 8, Nr. 3, 26. März 2013, S. e60403. bibcode:2013PLoSO...860403G. doi:10.1371/journal.pone.0060403. PMID 23555968. PMC 3608588 (freier Volltext).
- Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). In: Nature Genetics. 38, Nr. 11, November 2006, S. 1348–54. doi:10.1038/ng1896. PMID 17033623.
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J: Comprehensive mapping of long-range interactions reveals folding principles of the human genome. In: Science. 326, Nr. 5950, Oktober 2009, S. 289–93. bibcode:2009Sci...326..289L. doi:10.1126/science.1181369. PMID 19815776. PMC 2858594 (freier Volltext).
- Harewood L, Kishore K, Eldridge MD, Wingett S, Pearson D, Schoenfelder S, Collins VP, Fraser P: Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. In: Genome Biology. 18, Nr. 1, Juni 2017, S. 125. doi:10.1186/s13059-017-1253-8. PMID 28655341. PMC 5488307 (freier Volltext).
- Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, Shendure J: Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. In: Nature Biotechnology. 31, Nr. 12, Dezember 2013, S. 1119–25. doi:10.1038/nbt.2727. PMID 24185095. PMC 4117202 (freier Volltext).
- Schmitt AD, Hu M, Ren B: Genome-wide mapping and analysis of chromosome architecture. In: Nature Reviews. Molecular Cell Biology. 17, Nr. 12, Dezember 2016, S. 743–755. doi:10.1038/nrm.2016.104. PMID 27580841. PMC 5763923 (freier Volltext).
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR: Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. In: Nature Genetics. 46, Nr. 2, Februar 2014, S. 205–12. doi:10.1038/ng.2871. PMID 24413732.
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, Hughes JR: Multiplexed analysis of chromosome conformation at vastly improved sensitivity. In: Nature Methods. 13, Nr. 1, Januar 2016, S. 74–80. doi:10.1038/nmeth.3664. PMID 26595209. PMC 4724891 (freier Volltext).
- Hughes JR, Roberts N, McGowan S, Hay D, Giannoulatou E, Lynch M, De Gobbi M, Taylor S, Gibbons R, Higgs DR: Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. In: Nature Genetics. 46, Nr. 2, Februar 2014, S. 205–12. doi:10.1038/ng.2871. PMID 24413732.
- Jäger R, Migliorini G, Henrion M, Kandaswamy R, Speedy HE, Heindl A, Whiffin N, Carnicer MJ, Broome L, Dryden N, Nagano T, Schoenfelder S, Enge M, Yuan Y, Taipale J, Fraser P, Fletcher O, Houlston RS: Capture Hi-C identifies the chromatin interactome of colorectal cancer risk loci. In: Nature Communications. 6, Februar 2015, S. 6178. bibcode:2015NatCo...6.6178J. doi:10.1038/ncomms7178. PMID 25695508. PMC 4346635 (freier Volltext).
- Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. In: Nature. 502, Nr. 7469, Oktober 2013, S. 59–64. bibcode:2013Natur.502...59N. doi:10.1038/nature12593. PMID 24067610. PMC 3869051 (freier Volltext).
- Single-cell epigenomics: techniques and emerging applications. In: Nature Reviews. Genetics. 16, Nr. 12, Dezember 2015, S. 716–26. doi:10.1038/nrg3980. PMID 26460349.
- Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T: Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. In: Nature Genetics. 37, Nr. 1, Januar 2005, S. 31–40. doi:10.1038/ng1491. PMID 15608638.
- The second decade of 3C technologies: detailed insights into nuclear organization. In: Genes & Development. 30, Nr. 12, Juni 2016, S. 1357–82. doi:10.1101/gad.281964.116. PMID 27340173. PMC 4926860 (freier Volltext).
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W: Looping and interaction between hypersensitive sites in the active beta-globin locus. In: Molecular Cell. 10, Nr. 6, Dezember 2002, S. 1453–65. doi:10.1016/S1097-2765(02)00781-5. PMID 12504019.
- Cavalli G, Misteli T: Functional implications of genome topology. In: Nature Structural & Molecular Biology. 20, Nr. 3, März 2013, S. 290–9. doi:10.1038/nsmb.2474. PMID 23463314. PMC 6320674 (freier Volltext).
- Dekker J, Marti-Renom MA, Mirny LA: Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. In: Nature Reviews. Genetics. 14, Nr. 6, Juni 2013, S. 390–403. doi:10.1038/nrg3454. PMID 23657480. PMC 3874835 (freier Volltext).
- Guo Y, Xu Q, Canzio D, Shou J, Li J, Gorkin DU, Jung I, Wu H, Zhai Y, Tang Y, Lu Y, Wu Y, Jia Z, Li W, Zhang MQ, Ren B, Krainer AR, Maniatis T, Wu Q: CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. In: Cell. 162, Nr. 4, August 2015, S. 900–10. doi:10.1016/j.cell.2015.07.038. PMID 26276636. PMC 4642453 (freier Volltext).
- Regulation of disease-associated gene expression in the 3D genome. In: Nature Reviews. Molecular Cell Biology. 17, Nr. 12, Dezember 2016, S. 771–782. doi:10.1038/nrm.2016.138. PMID 27826147.
- Characterisation of deletions which affect the expression of fetal globin genes in man. In: Nature. 279, Nr. 5714, Juni 1979, S. 598–603. bibcode:1979Natur.279..598F. doi:10.1038/279598a0. PMID 450109.
- gamma-beta-Thalassaemia studies showing that deletion of the gamma- and delta-genes influences beta-globin gene expression in man. In: Nature. 283, Nr. 5748, Februar 1980, S. 637–42. doi:10.1038/283637a0. PMID 6153459.
- A functional screen for sonic hedgehog regulatory elements across a 1 Mb interval identifies long-range ventral forebrain enhancers. In: Development. 133, Nr. 4, Februar 2006, S. 761–72. doi:10.1242/dev.02239. PMID 16407397.
- Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. In: Nature Genetics. 48, Nr. 2, Februar 2016, S. 176–82. doi:10.1038/ng.3470. PMID 26656844.
- Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. In: Science. 346, Nr. 6215, Dezember 2014, S. 1373–7. doi:10.1126/science.1259037. PMID 25394790. PMC 4720521 (freier Volltext).
- Lajoie BR, van Berkum NL, Sanyal A, Dekker J: My5C: web tools for chromosome conformation capture studies. In: Nature Methods. 6, Nr. 10, Oktober 2009, S. 690–1. doi:10.1038/nmeth1009-690. PMID 19789528. PMC 2859197 (freier Volltext).
- Deng X, Ma W, Ramani V, Hill A, Yang F, Ay F, Berletch JB, Blau CA, Shendure J, Duan Z, Noble WS, Disteche CM: Bipartite structure of the inactive mouse X chromosome. In: Genome Biology. 16, Nr. 1, August 2015, S. 152. doi:10.1186/s13059-015-0728-8. PMID 26248554. PMC 4539712 (freier Volltext).
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. In: Cell. 159, Nr. 7, Dezember 2014, S. 1665–80. doi:10.1016/j.cell.2014.11.021. PMID 25497547. PMC 5635824 (freier Volltext).
- Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, Wang T: Exploring long-range genome interactions using the WashU Epigenome Browser. In: Nature Methods. 10, Nr. 5, Mai 2013, S. 375–6. doi:10.1038/nmeth.2440. PMID 23629413. PMC 3820286 (freier Volltext).
- Yardımcı GG, Noble WS: Software tools for visualizing Hi-C data. In: Genome Biology. 18, Nr. 1, Februar 2017, S. 26. doi:10.1186/s13059-017-1161-y. PMID 28159004. PMC 5290626 (freier Volltext).
- Genome-wide mapping and analysis of chromosome architecture. In: Nature Reviews. Molecular Cell Biology. 17, Nr. 12, Dezember 2016, S. 743–755. doi:10.1038/nrm.2016.104. PMID 27580841. PMC 5763923 (freier Volltext).
- Cavalli G, Misteli T: Functional implications of genome topology. In: Nature Structural & Molecular Biology. 20, Nr. 3, März 2013, S. 290–9. doi:10.1038/nsmb.2474. PMID 23463314. PMC 6320674 (freier Volltext).
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. In: Cell. 159, Nr. 7, Dezember 2014, S. 1665–80. doi:10.1016/j.cell.2014.11.021. PMID 25497547. PMC 5635824 (freier Volltext).
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. In: Cell. 159, Nr. 7, Dezember 2014, S. 1665–80. doi:10.1016/j.cell.2014.11.021. PMID 25497547. PMC 5635824 (freier Volltext).
- Yardımcı GG, Noble WS: Software tools for visualizing Hi-C data. In: Genome Biology. 18, Nr. 1, Februar 2017, S. 26. doi:10.1186/s13059-017-1161-y. PMID 28159004. PMC 5290626 (freier Volltext).
- Iterative correction of Hi-C data reveals hallmarks of chromosome organization. In: Nature Methods. 9, Nr. 10, Oktober 2012, S. 999–1003. doi:10.1038/nmeth.2148. PMID 22941365. PMC 3816492 (freier Volltext).
- Zambelli F, Pesole G, Pavesi G: Motif discovery and transcription factor binding sites before and after the next-generation sequencing era. In: Briefings in Bioinformatics. 14, Nr. 2, März 2013, S. 225–37. doi:10.1093/bib/bbs016. PMID 22517426. PMC 3603212 (freier Volltext).
- Bailey, S. D., Zhang, X., Desai, K., Aid, M., Corradin, O., Cowper-Sal·lari, R., … Lupien, M. (2015). ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nature Communications, 2, 6186. Retrieved from http://dx.doi.org/10.1038/ncomms7186
- Harewood L, Kishore K, Eldridge MD, Wingett S, Pearson D, Schoenfelder S, Collins VP, Fraser P: Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. In: Genome Biology. 18, Nr. 1, Juni 2017, S. 125. doi:10.1186/s13059-017-1253-8. PMID 28655341. PMC 5488307 (freier Volltext).
- Taberlay PC, Achinger-Kawecka J, Lun AT, Buske FA, Sabir K, Gould CM, Zotenko E, Bert SA, Giles KA, Bauer DC, Smyth GK, Stirzaker C, O'Donoghue SI, Clark SJ: Three-dimensional disorganization of the cancer genome occurs coincident with long-range genetic and epigenetic alterations. In: Genome Research. 26, Nr. 6, Juni 2016, S. 719–31. doi:10.1101/gr.201517.115. PMID 27053337. PMC 4889976 (freier Volltext).