Walsh-Modell
Das Walsh-Modell ist ein von Arthur Donald Walsh zuerst für Cyclopropan entwickeltes Modell, das auf dem Molekülorbital-Konzept der chemischen Bindung basiert.[1]
Vorgeschichte
Die Geometrie und Reaktivität von Cyclopropan ließen sich näherungsweise erklären durch das Baeyer-Modell und das Modell der gebogenen Bindungen (Bananenbindungen). Diese Modelle zeigen jedoch Schwachpunkte; die für Cyclopropan charakteristischen elektrophilen Additionsreaktionen lassen sich so nicht zufriedenstellend deuten. Daher schlug Walsh 1947 eine andere Erklärung vor.
Erstellung und Energieniveaus
Das Cyclopropan-Modell nach Walsh wird konstruiert, indem man drei trigonale CH2-Gruppen so zusammenbringt, dass die durch die beiden Wasserstoffatome und das Kohlenstoffatom der Methylengruppen gebildete Ebene senkrecht zur Ebene des Dreirings steht. In der Ebene des Dreirings liegen dann die 2pz-Atomorbitale (2pz-AO) der sp2-hybridisierten CH2-Gruppe und ein „Lappen“ des sp2-Hybridorbitals.
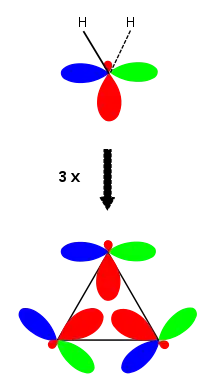
Oben: sp2-hybridisierte CH2-Gruppe
Unten: die C-C-Bindungen in Cyclopropan
Die Molekülorbitale (MOs) des Cyclopropans werden aus den Orbitalen der Teilstücke (–CH2–) durch Linearkombination gebildet. Drei MOs des Cyclopropans erhält man durch Linearkombination der drei ins Innere des Dreirings weisenden Lappen der sp2–Hybridorbitale (φ1, φ2 und φ3). Drei weitere Molekülorbitale erhält man durch Linearkombination der 2pz-AOs (Φ1, Φ2 und Φ3).
Aus diesen beiden Sätzen der Basisorbitale φ1, φ2 und φ3 (sp2-Typ), sowie Φ1, Φ2 und Φ3 (2p-Typ), erhält man durch Linearkombination (Säkulardeterminante 3. Ordnung) folgende Wellenfunktionen der drei MOs:



sowie





Mit den in der organischen Chemie üblichen vereinfachten Darstellungen – die „Ausdehnung“ der Orbitale bleibt unberücksichtigt – lassen sich diese Molekülorbitale anschaulich machen.
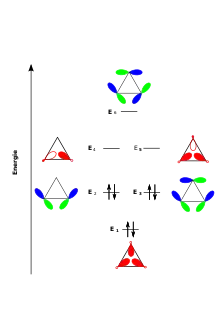
Unten die energieärmeren bindenden MOs, die besetzt sind, oben die antibindenden MOs
Die Molekülorbitale Ψ2 und Ψ3, sowie ψ2 und ψ3 sind jeweils energiegleich („entartet“, siehe nebenstehendes Bild). Von den sechs eingebrachten Elektronen besetzen 2 Elektronen das MO ψ1. Ihre Aufenthaltswahrscheinlichkeit ist das „dreilappige Gebiet“ im Inneren des Cyclopropanringes. Diese Elektronen sind am festesten gebunden. Die MOs ψ2 und ψ3 sind nicht besetzt; sie sind ziemlich stark antibindend (entlang der C-C-Bindungen ist die Zahl der Knoten größer als die Zahl der bindenden Wechselwirkungen). Beim Satz der MOs Ψ1, Ψ2 und Ψ3 ist die Situation gerade umgekehrt wie bei den soeben behandelten MOs: Ψ1 ist antibindend (dreimal Vorzeichenwechsel entlang der C-C-Bindungen), während Ψ2 und Ψ3 insgesamt bindend sind. Ψ2 und Ψ3 sind daher von niedrigster Energie; diese beiden MOs werden von 4 Elektronen besetzt. Die Aufenthaltswahrscheinlichkeit dieser Elektronen ist an der Peripherie des Dreiringes besonders hoch; dies sind die reaktiveren „Valenzelektronen“ des Cyclopropans. Die Bindungen in den Molekülorbitalen Ψ2 und Ψ3 haben eine gewisse Ähnlichkeit mit den π-Bindungen im Ethen. Sie haben sowohl σ- als auch π-Charakter. Die Größe der Überlappung liegt zwischen dem niedrigen Wert für die reine p-π-Überlappung im Ethen und der starken σ-Überlappung der sp3-Orbitale in „gesättigten“ Kohlenwasserstoffen.
Die berechneten Energien der Molekülorbitale und ihre Besetzung durch Elektronen können in einem schematischen Bild dargestellt werden.
Im Rahmen des Grenzorbital-Konzeptes der chemischen Reaktivität (HOMO/LUMO) sind beim Cyclopropan die Molekülorbitale Ψ2 und Ψ3 die energetisch höchsten besetzten Orbitale (HOMOs). Sie gehen Wechselwirkungen mit Protonen und den LUMOs von elektrophilen Reagentien ein.
Das Walsh-Modell erlaubt somit sowohl eine π-Bindung, die das kleine Ringmolekül trotz ungünstigen Winkels zusammenhält, als auch ein besetztes da relativ energieärmeres p-Orbital, das die Reaktivität des Ringmoleküls erklärt.
Schwachpunkte
Obwohl das Walsh-Modell unter organischen Chemikern sehr beliebt war/ist,[2] hat es Schwächen, auf die mehrfach hingewiesen wurde.[3][4][5] Die Orbitale nach Walsh sind den Orbitalen nach dem Förster-Coulson-Moffitt-Modell nicht äquivalent. Für die detaillierte Kritik muss auf die Originalliteratur verwiesen werden.
Weitere Entwicklung
Hückel-Näherung, Möbius-Aromaten und Hellmann-Feynman-Theorem sind weitere Konzepte, die in die Thematik und die Weiterentwicklung dieses Modells eingeflossen sind.[6][7] Erst 1986 wies Dewar bei der Betrachtung des Molekülorbitals auf den für Aromaten mit ihrem für delokalisierte Doppelbindungen typischen σ-Anteil hin, der eine Anomalie der Spannungsenergie des Cyclopropans, das kein Aromat ist, dennoch begründen kann. Berechnungen der Laplace-Verteilung der Elektronen bestätigten ihre höhere Dichte inmitten der drei Atomkerne.[8]
Literatur
- Martin Klessinger: Elektronenstruktur organischer Moleküle. Verlag Chemie, Weinheim u. a. 1982, ISBN 3-527-25925-2, S. 101ff, S. 264–266.
- A. D. Walsh: Structures of Ethylene Oxide and Cyclopropane. In: Nature. Band 159, Nr. 4031, 1947, S. 165–165, doi:10.1038/159165a0.
Einzelnachweise
- A. D. Walsh: The structures of ethylene oxide, cyclopropane, and related molecules. In: Transactions of the Faraday Society. 45, 1949, S. 179–190, doi:10.1039/TF9494500179.
- Siehe z. B. H. R. Christen, F. Vögtle: Organische Chemie – Von den Grundlagen zur Forschung. Bd. 2, 1. Auflage. Otto Salle Verlag, Frankfurt a. Main 1990, ISBN 3-7935-5398-1, S. 284–285.
- E. Honegger, E. Heilbronner, A. Schmelzer In: Nouveau Journal de Chimie. 6, 519 (1982).
- K. B. Wiberg: Structures, energies and spectra of cyclopropanes. In: Zvi Rappoport (Hrsg.): Patai's Chemistry of Functional Groups. The Chemistry of the Cyclopropyl Group. Wiley, Chichester u. a. 1987, S. 1–4.
- D. Cremer, E. Kraka, K. J. Szabo: General and Theoretical Aspects of the Cyclopropyl Group. In: Zvi Rappoport (Hrsg.): Patai's Chemistry of Functional Groups. The chemistry of the cyclopropyl group. Band 2, Wiley, Chichester 1995, ISBN 0-471-94074-7. doi:10.1002/9780470682531.pat0028.
- Wolfgang W. Schoeller, Thomas Dabisch: Substituent effects on the bonding properties in cyclotriphosphanes and related compounds. Polarization of Hückel versus Möbius orbitals. In: Journal of the Chemical Society, Dalton Transactions. Band 0, Nr. 11, 1983, S. 2411–2413, doi:10.1039/DT9830002411.
- Vincenzo Barone, Sándor Fliszár: Theoretical energies of representative carbon-carbon bonds. In: International Journal of Quantum Chemistry. Band 55, Nr. 6, 1995, S. 469–476, doi:10.1002/qua.560550605.
- Francis A. Carey, Richard J. Sundberg: Advanced Organic Chemistry: Reactions and synthesis. 2005, S. 85ff (eingeschränkte Vorschau in der Google-Buchsuche).