Mevalonatkinase-Mangel
Der Mevalonatkinase-Mangel ist eine sehr seltene angeborene Stoffwechselstörung mit beeinträchtigter Biosynthese von Cholesterin und Isoprenoiden.[1][2]
| Klassifikation nach ICD-10 | |
|---|---|
| E88.8 | Sonstige näher bezeichnete Stoffwechselstörungen |
| E85.0 | Nichtneuropathische heredofamiliäre Amyloidose |
| ICD-10 online (WHO-Version 2019) | |
Synonyme sind: Mevalonazidämie; Mevalonazidurie; ATP-Mevalonat-5-Phosphotransferase-Mangel; HIDS; Hyper-IgD-Syndrom; Hyperimmunglobulinämie D mit periodischem Fieber; Hyperimmunglobulinämie D-Syndrom; Mevalonatkinase-Mangel, partieller; Mevalonatkinase-Mangel, vollständiger
Die Bezeichnung umfasst[3] das Hyper-IgD-Syndrom mit Rückfallfieber (HIDS)[4] und die Mevalonazidurie (MVA).[5][6]
Der Mevalonatkinase-Mangel ist ein uneinheitliches Krankheitsbild mit unterschiedlichen klinischen Erscheinungen. Das Spektrum dieser Symptome reicht von wenig ausgeprägt beim Hyperimmunoglobulinämie D-Syndrom (HIDS) bis zur schwereren Erkrankung, der Mevalonazidurie (MVA).
Die Erstbeschreibung der Hyperimmunglobulinämie D mit Rückfallfieber stammt aus dem Jahre 1984 durch den niederländischen Internisten JosW.M. van der Meer und Mitarbeiter[7], die der Mevalonazidurie stammt aus dem Jahre 1986 durch Georg Hoffmann, Kenneth M. Gibson, Ira K. Brandt und Mitarbeiter.[8]
Verbreitung
Die Häufigkeit wird mit unter 1 zu 1.000.000 angegeben, bislang wurde über mindestens 300 Betroffene, davon 30 mit MVA und 180 mit HIDS, berichtet. Die Vererbung erfolgt autosomal-rezessiv.[5]
Ursache
Den Erkrankungen liegen Mutationen im MVK-Gen auf Chromosom 12 Genort q24.11 zugrunde, welche zu unterschiedlich stark verminderter Aktivität der Mevalonatkinase führen.[9][10] Fehlt das Enzym nahezu ganz, kommt es zur schwereren Form MVA.[5]
Klinische Erscheinungen
Klinische Kriterien sind:[1][5][11]
- Krankheitsbeginn als Neugeborenes, Kleinkind oder im Kindesalter
bei HIDS:
- meist im ersten Lebensjahr wiederholte Fieberattacken mit Erbrechen und Diarrhoe, 3 bis 7 Tage anhaltend, im Abstand weniger Wochen auftretend, im späteren Alter seltener werdend
- Gelenkschmerzen, geschwollene Lymphknoten, Kopfschmerzen und Hautveränderungen
Hinzu können neurologische Auffälligkeiten, Geistige Behinderung, Ataxie, Sehrstörungen und Krampfanfälle kommen.
Bei MVA:
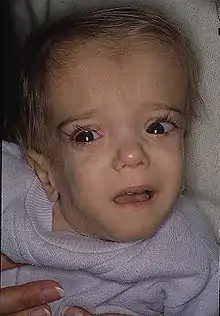
- Gesichtsauffälligkeiten wie Hypertelorismus, Balkonstirn, lange Wimpern und dreieckige Gesichtsform
- statomotorische Entwicklungsverzögerung
- Gedeihstörung
- kleinhirnbedingte Ataxie
- Anämie
- Katarakt mit fortschreitender Einschränkung des Sehvermögens
- rezidivierende Fieberattacken mit Lymphknotenschwellungen, Hepatosplenomegalie, Gelenkschmerzen und Exanthemen.
Bei Erwachsenen kann es als Komplikation zu Amyloidose, Verwachsungen im Bauchraum und selten zu Kontrakturen kommen.[5]
Diagnose
Die Diagnose ergibt sich aus den klinischen Befunden, der Humangenetischen Untersuchung und dem Nachweis des MVK-Mangels. Der IgD-Spiegel im Serum ist – außer in den ersten Lebensjahren – oft erhöht, in 80 % ist auch der IgA-Spiegel erhöht, der Cholesterin-Spiegel kann erniedrigt sein. Im Urin ist die Ausscheidung der Mevalonsäure stark erhöht.[1][5]
Differentialdiagnostik
Abzugrenzen sind:[5]
Therapie
Zur Behandlung bei HIDS kann Simvastatin erfolgreich sein.[5]
Literatur
- J. Jeyaratnam, J. Frenkel: Management of Mevalonate Kinase Deficiency: A Pediatric Perspective. In: Frontiers in immunology. Band 11, 2020, S. 1150, doi:10.3389/fimmu.2020.01150, PMID 32582214, PMC 7289972 (freier Volltext) (Review).
- A. R. Correa, N. Gupta, N. Bagri, P. Vignesh, S. Alam, S. Yamaguchi: Mevalonate Kinase Deficiency as Cause of Periodic Fever in Two Siblings. In: Indian pediatrics. Band 57, Nummer 2, 02 2020, S. 180–181, PMID 32060250.
- G. Horneff: [Effectiveness of interleukin-1 inhibiton for mevalonate kinase deficiency]. In: Zeitschrift fur Rheumatologie. Band 73, Nummer 5, Juni 2014, S. 398, PMID 25057522.
Einzelnachweise
- Bernfried Leiber (Begründer): Die klinischen Syndrome. Syndrome, Sequenzen und Symptomenkomplexe. Hrsg.: G. Burg, J. Kunze, D. Pongratz, P. G. Scheurlen, A. Schinzel, J. Spranger. 7., völlig neu bearb. Auflage. Band 2: Symptome. Urban & Schwarzenberg, München u. a. 1990, ISBN 3-541-01727-9.
- J. Mancini, N. Philip, B. Chabrol, P. Divry, M. O. Rolland, N. Pinsard: Mevalonic aciduria in 3 siblings: a new recognizable metabolic encephalopathy. In: Pediatric neurology. Band 9, Nummer 3, 1993 May-Jun, S. 243–246, doi:10.1016/0887-8994(93)90095-t, PMID 8352861.
- Mevalonatkinase-Mangel. In: Orphanet (Datenbank für seltene Krankheiten).
- Hyperimmunglobulinämie D mit Rückfallfieber. In: Orphanet (Datenbank für seltene Krankheiten).
- Mevalonazidurie. In: Orphanet (Datenbank für seltene Krankheiten).
- G. F. Hoffmann, D. Haas: Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. In: Orphanet Journal of Rare Diseases. 2006; 1, S. 13.
- J. W. van der Meer, J. M. Vossen, J. Radl, J. A. van Nieuwkoop, C. J. Meyer, S. Lobatto, R. van Furth: Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. In: The Lancet. Band 1, Nummer 8386, Mai 1984, S. 1087–1090, doi:10.1016/s0140-6736(84)92505-4, PMID 6144826.
- G. Hoffmann, K. M. Gibson, I. K. Brandt, P. I. Bader, R. S. Wappner, L. Sweetman: Mevalonic aciduria–an inborn error of cholesterol and nonsterol isoprene biosynthesis. In: The New England Journal of Medicine. Band 314, Nummer 25, Juni 1986, S. 1610–1614, doi:10.1056/NEJM198606193142504, PMID 3012338.
- Hyper-IgD syndrome. In: Online Mendelian Inheritance in Man. (englisch)
- Mevalonic aciduria. In: Online Mendelian Inheritance in Man. (englisch)
- NORD Rare Diseases